
Abstract
Beta-ketothiolase (mitochondrial acetoacetyl-CoA thiolase, T2) deficiency (OMIM #203750, *607809) is an inborn error of metabolism that affects isoleucine catabolism and ketone body metabolism. This disorder is clinically characterized by intermittent ketoacidotic crises under ketogenic stresses. In addition to a previous 26-case series, four series of T2-deficient patients were recently reported from different regions. In these series, most T2-deficient patients developed their first ketoacidotic crises between the ages of 6 months and 3 years. Most patients experienced less than three metabolic crises. Newborn screening (NBS) for T2 deficiency is performed in some countries but some T2-deficient patients have been missed by NBS. Therefore, T2 deficiency should be considered in patients with severe metabolic acidosis, even in regions where NBS for T2 deficiency is performed. Neurological manifestations, especially extrapyramidal manifestations, can occur as sequelae to severe metabolic acidosis; however, this can also occur in patients without any apparent metabolic crisis or before the onset of metabolic crisis.
Similar content being viewed by others


Introduction
Beta-ketothiolase (mitochondrial acetoacetyl-CoA thiolase, T2, EC 2.3.1.9) deficiency (OMIM #203750, *607809) was first described as an inborn error of isoleucine catabolism in 1971 [1,2,3]. Later this disorder was revealed to be a defect in mitochondrial acetoacetyl-CoA thiolase activity that affects both isoleucine catabolism and ketolysis [4]. Hence, this disorder is classified as an organic aciduria or as a defect in ketolysis. The disorder is clinically characterized by intermittent ketoacidotic episodes [5,6,7]. Our group has intensively studied T2 deficiency from both clinical and molecular perspectives [3, 8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. A cohort of 26 cases was previously reported in 2001 [25]. In 2017, four more cohorts of 26, 41, 32, and 10 cases from different areas and ethnicities were reported [41, 42, 46, 47]. This number of cases may be sufficient to understand the clinical manifestation of the disorder. Here, we review the clinical manifestation of T2 deficiency, mainly in patients from the five reported cohorts and we discuss what remains to be solved.
We will especially focus on newborn screening (NBS) and neurological manifestation of the disorder. T2 deficiency is included in newborn screening in some countries, although there are several reports of false-negative results [46, 48,49,50,51], including in Japanese cases [3, 15, 28]. Neurological manifestation/complication in T2 deficiency has been regarded as a sequela of severe metabolic acidosis; however, several patients have developed neurological problems without apparent severe metabolic acidosis [9, 13, 25, 26, 31, 32, 35, 38, 46, 47, 52].
Pathophysiology
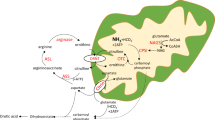
T2 catalyzes the interconversion of acetyl-CoA and acetoacetyl-CoA in mitochondria of both liver and extrahepatic tissues (Fig. 1) [5]. During hepatic ketogenesis, the T2 reaction in the direction of acetoacetyl-CoA formation from abundant acetyl-CoA is favored. In extrahepatic tissues, T2 catalyzes predominantly acetoacetyl-CoA cleavage to form acetyl-CoA. Because ketoacidosis is the main feature of T2 deficiency, its function in ketolysis is physiologically more important than its role in hepatic ketogenesis. In T2-deficient liver, another thiolase, mitochondrial medium-chain 3-ketoacyl-CoA thiolase (T1), which mainly functions to cleave medium-chain 3-ketoacyl-CoA during fatty acid beta-oxidation, may compensate for T2 deficiency [53].

Overview of ketone body metabolism and isoleucine catabolism. T2 is involved in isoleucine catabolism, ketogenesis in the liver, and ketolysis in extrahepatic tissues. 2M3HB 2-methyl-3-hydroxybutyrate, 2M3HB-CoA 2-methyl-3-hydroxybutyryl-CoA, 2M3HBD 2-methyl-3-hydroxybutyrate dehydrogenase, 2MAcAc 2-methylacetoacetate, 2MAcAc-CoA 2-methylacetoacetyl-CoA, 3HB 3-hydroxybutyrate, 3HBD 3-hydroxybutyrate dehydrogenase, Ac-CoA acetyl-coenzyme A, AcAc Acetoacetate, AcAc-CoA acetoacetyl-CoA, ACs acylcarnitines, CoA coenzyme A, HMG-CoA 3-hydroxy-3-methylglutaryl-CoA, HMGCL HMG-CoA lyase, HMGCS2 mitochondrial HMG-CoA synthase, SCOT succinyl-CoA:3-oxoacid CoA transferase, T1 mitochondrial medium-chain 3-ketoacyl-CoA thiolase, T2 mitochondrial acetoacetyl-CoA thiolase, TCA tricarboxylic acid, TIG tiglylglycine, UOAs urinary organic acids
T2 also catalyzes 2-methylacetoacetyl-CoA cleavage into acetyl-CoA and propionyl-CoA in isoleucine catabolism [54]. T2 deficiency results in accumulation of isoleucine-catabolic intermediates, such as 2-methylacetoacetyl-CoA, 2-methyl-3-hydroxybutyryl-CoA and tiglyl-CoA. Urinary elevated excretion of 2-methylacetoacetate (2MAcAc), 2-methyl-3-hydroxybutyrate (2M3HB), and tiglylglycine (TIG) is characteristic for T2 deficiency, while elevations of C5-OH carnitine (2-methyl-3-hydroxybutyryl-carnitine) and C5:1 carnitine (tiglylcarnitine) are characteristic in blood acylcarnitine analysis. These chemical features are in accord with the fact that T1 has little affinity for 2-methylacetoacetyl-CoA as a substrate. Of note, these chemical features, when present, are highly suggestive of T2 deficiency but their absence, especially during an asymptomatic period, does not absolutely rule out T2 deficiency [3, 28].
The T2 enzyme is a homotetramer of 41 kDa. The T2 gene (Acetyl Coenzyme A Acetyltransferase 1, ACAT1) is located on chromosome 11q22.3-23.1 and includes 12 exons that span ~27 kb. At Gifu University, we have identified more than 80 T2 gene mutations in more than 120 patients. There are some common mutations in specific populations. For example, p.R208* and c.1006-1g>c were identified in nearly 70 and 20%, respectively, of mutant alleles in the Vietnamese population [41]. p.M193R was identified in nearly 50% of mutant alleles in the Indian population [42]. Other common mutations identified in at least four independent T2-deficient families are p.G152A, p.N158D, p.N158S, p.G183R, p.Q272X, c.828+1g>t, p.T297M, and c.1163+2t>c. There is a significant biochemical-genotype correlation, but no apparent phenotype–genotype correlation. Transient expression analysis of mutant cDNAs showed some mutations retain significant residual T2 activities. Such mutations are designated as “mild” mutations. In this review, patients with mild genotypes are defined as patients who have “mild” mutations in at least one of two mutant alleles.
Clinical manifestation
A series of 26 cases was previously reported in 2001 [25]. In 2017, other series of 26 cases from France [47], 41 cases from Vietnam [41], 32 mostly from Turkey, The Netherlands, and Germany [46], and 10 from India [42] were reported. Hence in total, 135 T2-deficient patients are described in these five reports. The study of 2001 clearly described the clinical manifestation of T2-deficient patients and is still the most informative in some aspects (Table 1). The latest four reports provide some unique information. The series from Vietnam provides typical clinical information for patients who have two null mutations (severe genotype), because most patients had a combination of two null mutations, R208X and c.1006-1g>c [41]. The series from France focused on neurological complications in T2 deficiency [47]. We will discuss neurological manifestations together with these data later. The series from Turkey, The Netherlands and Germany [46], and from France [47] provide information of patients who were diagnosed in early childhood and who were more than 10 years-old at the time of study.
Onset
Figure 2a shows the onset of symptoms for the 135 patients listed in the five series. Most patients developed their first ketoacidotic crisis between 6 and 36 months of age with the peak onset at 6–11 months of age. Neonatal onset of T2 deficiency is very rare and only two patients had neonatal onset. The early neonatal period is one of catabolic conditions and is a major period for the onset peak of typical organic acidemia, such as isovaleric acidemia, propionic acidemia, methylmalonic acidemia, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) lyase deficiency as well as another ketolytic defect, succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency [6]. Only three patients had onset after 6 years of age. Fourteen percent of patients did not have any metabolic episodes; among them 11 patients were diagnosed through NBS or familial analysis in infancy and eight patients were diagnosed after 1 year of age by familial analyses. Figure 2b shows the onset age of 57 and 12 patients with confirmed severe and mild genotypes, respectively. The peak of onset age was 6–11 and 12–17 months in patients with severe and mild genotypes, respectively, although the number of patients with mild genotype was small. In most cases, the first ketoacidotic episode followed an intercurrent illness, especially gastroenteritis or respiratory disease in association with fasting and catabolic conditions. The onset of T2 deficiency was common in late infancy and early childhood because children usually experience their first episodes of infection during this period. In one case, excessive protein intake on the day preceding the episode was reported. In T2 deficiency, catabolic conditions appear to confer a higher risk than that of protein overload.

Onset of first ketoacidotic crises. a Onset of first metabolic crisis in all patients in the five series are shown. The vertical and horizontal axes indicate number of patients and their age in months, respectively. I indicates that patients were identified as having T2 deficiency before 1 year of age by newborn screening or familial screening; II indicates that patients were identified as having T2 deficiency after 1 year of age by familial analysis. b Onset of first metabolic crisis in 57 and 12 patients with confirmed severe and mild genotypes, respectively. The vertical and horizontal axes indicate number of patients and their age in months, respectively
Metabolic profiles in the first crises
Metabolic profiles are available for the Vietnamese series [41]. Most patients belong to the severe genotype; therefore, metabolic profiles are expected to be typical. Metabolic acidosis was in general severe. Blood pH and HCO3 ranged between 6.80–7.26 (mean 7.02) and 0–10.7 mmol/L (mean 3.2), respectively. In general, during metabolic acidosis, blood glucose levels vary, ranging between 1.7 and 23.3 mmol/L (mean 7.51). Hyperglycemia (>7.8 mmol/L) was observed in 28% of patients and hypoglycemia (<2.5 mmol/L) was present in 8% of patients. Hyperglycemia mimicking diabetic ketoacidosis is sometimes observed in another ketolytic defect (SCOT deficiency) [55] and in other organic acidurias [56,57,58]. Such hyperglycemia may be because of a stress reaction. Hyperammonemia (>170 μmol/L) presented in 23% of patients. However, the highest level of ammonia was 307 μmol/L. This is quite different from other organic acidemias. Hence, among 39 symptomatic patients only four needed hemodialysis, although mechanical ventilation was used in 16 patients.
Death
Among 135 patients, seven patients died of a severe ketoacidotic crisis or from sequelae of a severe crisis. In the first series, only GK16 died of sequelae of the first crisis [25]. She suffered a brain hemorrhage because of hypernatremia caused by excessive sodium bicarbonate use during the crisis. In the Vietnamese series, five patients died (two of them at the first crisis, two of them at the second crisis, and one patient died because of neurological sequelae following the first crisis) [41]. In the Indian series, one patient (GK108) died at the first crisis [8], and one patient died at the second crisis in the Turkish and German series [46]. The three patients above who died at the second metabolic crisis were diagnosed as T2 deficiency at the first metabolic crises; therefore, death could have been avoided.
Frequency of ketoacidotic crises
From the five series of T2-deficient patients, 39 were followed up for more than 10 years and the frequency of ketoacidotic crises in these patients was recorded (Fig. 3). About 34% of patients experienced only one metabolic crisis, which led to the diagnosis. This means that early diagnosis and management may prevent the development of severe subsequent metabolic crises. Most patients developed ketoacidotic crises only three times or less. Seven patients had no episodes. Information of the latest crises was available in two series. In the Vietnamese series, all metabolic crises occurred within 5 years of age [41], whereas in 26 cases reported in 2001, five patients developed severe metabolic crises between 5 and 10 years of age [25]. It should be noted that four of these five patients were diagnosed as T2 deficiency in their last crisis, when they were older than 5 years. Hence, after confirming the diagnosis of T2 deficiency, most metabolic crises occurred before 5 years of age. Even in a fasting test in normal children, blood ketone body levels are much lower at the age of 7–15 than at the age of 1–7 years [59]. This may mean ketone bodies are less important in older children for the maintenance of blood glucose levels compared with younger children and may explain why ketoacidotic events become less frequent with age in T2 deficiency.

Frequency of ketoacidotic crises. Frequency of metabolic crises in patients who were more than 10 years of age at last follow-up
Diagnosis
The main clinical manifestation of T2 deficiency is severe metabolic acidosis; therefore, urinary organic acid analysis is the most applicable second line metabolic analysis. T2 deficiency is chemically diagnosed by the presence of TIG, 2M3HB, and 2MAcAc from the isoleucine-catabolic pathway. 2MAcAc is unstable and can be difficult to detect. A combination of TIG and 2M3HB without 2MAcAc is characteristic for 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (2M3HBD) deficiency [60, 61]. This disorder is now called HSD10 disease and is an important differential diagnosis for T2 deficiency (Fig. 1). TIG was not detected in seven of the 26 patients in Table 1 even during acute crises; six of these patients were classified as mild genotype. Hence, the absence of TIG does not exclude T2 deficiency.
Among eight Japanese patients (Table 2), only one (GK01) had a severe genotype, the others being classified as mild genotype [3, 28]. These patients with mild genotype presented with severe ketoacidosis that was typical for patients with severe genotype, but their urinary organic acid and blood acylcarnitine profiles, especially during asymptomatic conditions, differ from those of typical severe genotype patients. In mild genotype patients, tiglylglycine may not be detected by urine organic acid analysis even during metabolic crisis. Between episodes, 2-methyl-3-hydroxybutyrate may also be only slightly elevated or even undetectable. Similarly, blood acylcarnitine analysis may be normal during non-episodic periods and even during acute episodes of metabolic decompensation in some T2-deficient patients with mild genotype (see Newborn screening for T2 deficiency).
Enzyme assays or mutation analysis is essential to confirm T2 deficiency. There are several kinds of enzyme assay. Measurement of potassium-ion activated acetoacetyl-CoA thiolase activity using fibroblasts is the gold standard method [62, 63]. Lymphocytes (peripheral blood mononuclear cells) can also be used but contamination with red blood cells may affect this assay. A 2-methylacetoacetyl-CoA thiolase specific assay was also reported [54] but this substrate is not commercially available. A coupling assay with tiglyl-CoA [64] cannot distinguish T2 deficiency from 2M3HBD deficiency (HSD10 disease).
Mutation analysis using genomic PCR and direct sequencing is a standard method for diagnosing T2 deficiency [22]. However cDNA analysis, multiplex ligation-dependent probe amplification, mutant cDNA expression analysis followed by enzyme assay, and a mini-gene splicing assay may be necessary to confirm whether an identified novel variant is a causative mutation or not [31, 32, 43].
Management
Management is divided into that for acute crisis and for non-episodic conditions [65].
Treatment of acute crisis
Even when the patient shows normoglycemia, intravenous glucose should be given to maintain blood glucose levels in the upper normal range; this is important to suppress ketogenesis. Insulin, together with sufficient glucose, can be used to further suppress ketosis in severe episodes, with frequent blood glucose monitoring. Treatment of metabolic acidosis using sodium bicarbonate is controversial. Minimal usage of sodium bicarbonate is recommended. In general, hemodialysis is not necessary if ketogenesis is successfully suppressed. Among Vietnamese 41 patients, hemodialysis was performed for only four patients [41].
Management in non-episodic conditions
It is important to avoid severe metabolic crises. For this purpose, fasting avoidance is the basis of long-term management. During ketogenic stressors, such as vomiting, appetite loss, or infection, sufficient oral glucose supplementation is essential to avoid ketoacidotic crises. Episodes associated with moderate or high urine ketones should be treated by intravenous glucose infusion. Mild dietary protein restriction (1.5–2 g/kg/day) is a fundamental and reasonable treatment to reduce isoleucine load. This is discussed later (neurological manifestation). Patients should also restrict excess fat intake; a ketogenic diet is contraindicated. l-carnitine supplementation may be considered, if patients have low free carnitine levels.
Newborn screening for T2 deficiency
T2 deficiency is a suitable disorder for NBS from a clinical perspective. This disorder rarely has neonatal onset and most patients develop ketoacidotic crises during 6 months to 3 years of age. In most patients, the first ketoacidotic crisis is the most severe and is associated with their prognosis. Such severe metabolic crises could be avoided if parents knew how to manage the condition. However, false-negative results appear to be inevitable in NBS for T2 deficiency.
We predicted in 2003 that NBS might miss some T2-deficient patients based on acylcarnitine profiles in Japanese patients [28]. Later, we also reported that levels of C5OH and C5:1 were within normal ranges in acylcarnitine profiles even during acute metabolic crises in two T2-deficient cases, GK77 and his brother GK77b [3]. As shown in Table 2, in Japan, most T2-deficient patients identified thus far, have at least one “mild” mutation, which retains significant residual T2 activity. In these patients, metabolites from isoleucine catabolism in blood acylcarnitine and urinary organic acid analyses were much lower than that in typical T2-deficient patients with two null mutations. Hence, we expected that NBS might miss such patients with mild mutations. This is why T2 deficiency is classified among secondary target diseases that are not necessarily screened in NBS in Japan. However, in our opinion, T2 deficiency should be included in NBS; even if routine NBS misses some T2-deficient patients, some will still be detected.
False-negative results in NBS were reported in eight patients from seven families [48,49,50,51]. Table 3 shows false-negative NBS results in these patients. Different cutoff values were used in different laboratories. From July 1997 to July 2005, NBS in North Carolina, USA, identified two T2-deficient patients through elevated C5:1 [51]. During this period, one T2-deficient patient was missed by NBS and later at 15 months of age this patient developed ketoacidosis. In a similar NBS program in Minnesota, USA, from January 2001 to November 2010, one case was identified and two sibling cases were missed [48]. The elder brother developed severe metabolic acidosis at the age of 10 months. He was suspected as having T2 deficiency by urinary organic acid profile and his diagnosis was confirmed by enzyme assay and mutation detection. Even during acute crises, his plasma C5-OH and C5:1 levels were normal, similar with cases GK77 and GK77b [3]. In the NBS program of New South Wales, Australia, from 1998 to 2012, two patients with T2 deficiency were missed using a screening marker of C5-OH [50]. A Boston group also reported two T2-deficient patients missed by NBS [49]. These patients developed normally before their first metabolic crises and manifested neurological complications after the crises. Hence, it is important that T2 deficiency should be considered for a patient with severe metabolic acidosis even in regions where T2 deficiency is included in NBS target diseases.
NBS studies have shown the incidence of T2 deficiency in some regions. The incidence of T2 deficiency in North Carolina, USA, from 1997 to 2005 was nearly 1 per 313,000 newborns [51], whereas that in Minnesota, USA, from January 2001 to November 2010 was 1 per 232,000 newborns [48]. No T2-deficient patients were identified in 3.36 million Japanese newborns up to 2015 by tandem mass spectrometry NBS including pilot NBS [66]. Furthermore, no T2-deficient patients missed by NBS were detected in Japan.
In most newborn screening centers in Japan, screening criteria for T2 deficiency is that both C5-OH and C5:1 are elevated to more than 1.0 and 0.05 nmol/mL, respectively. On its own, C5-OH elevated to more than 1.0 nmol/mL is used as a screening marker for methylcrotonyl-CoA carboxylase deficiency, multiple carboxylase deficiency and HMG-CoA lyase deficiency. Differential diagnosis for these conditions is made by urinary organic acid analysis. Hence, if a T2-deficient patient presents with elevated C5-OH, careful detection of 2MAcAc in a urinary organic acid profile leads to the diagnosis of T2 deficiency. Increased C5:1 is a specific marker for T2 deficiency and HSD10 deficiency (see next paragraph). If C5:1 is used independently as a screening marker for T2 deficiency and HSD10 disease with a lower cutoff level than the present value, more T2-deficient patients may be positively screened, although the C5:1 level is associated with some catabolic conditions.
2M3HBD deficiency (HSD10 disease) is the most important differential diagnosis of T2 deficiency in NBS-screened patients. 2M3HBD deficiency is a rare X-linked neurodegenerative condition caused by abnormalities in the HSD17B10 gene [60, 61, 66]. HSD10 protein is a multifunctional protein and functions as 2M3HBD, mitochondrial 17beta-hydroxysteroid dehydrogenase and is one of the three components of mitochondrial RNase P. The classical infantile form of HSD10 disease is characterized by a progressive neurodegenerative course with retinopathy and cardiomyopathy, although HSD10 disease has broad clinical heterogeneity. Because this disorder affects one step upstream from T2 deficiency in isoleucine catabolism (Fig. 1), a profile of elevated C5:1 and C5-OH is characteristic for both HSD10 disease and T2 deficiency. Hence, theoretically, this disorder is also screened for by the same criteria as T2 deficiency in NBS. To our knowledge, no HSD10 disease patient has been positively screened by NBS thus far. A profile of elevated urinary TIG and 2M3HB is also a shared characteristic for these two disorders. The only difference is the absence of 2MAcAc in HSD10 disease. However, it is sometimes difficult to detect 2MAcAc in T2 deficiency because of 2MAcAc instability. Hence, if a male baby is positively screened for T2 deficiency, this baby should be carefully examined for the possibility of both disorders. In one 2M3HBD deficient patient, pilot NBS at 5 days of age showed C5:1 levels of 0.07 nmol/mL and C5-OH levels of 0.29 nmol/mL. Notably, C5:1 was elevated, whereas C5-OH was within its cutoff limit in this 2M3HB deficient patient [67]. Such high C5:1 levels were not observed in more than 10,000 newborns in Gifu, Japan (unpublished observation). Thus, these data suggest that some T2 deficiency and HSD10 disease could be positively screened by NBS. Further analysis is necessary.
Neurological manifestation
It is well-known that neurological complications, such as developmental delay, ataxia, myoclonus, and other extrapyramidal symptoms are caused as sequelae of severe metabolic crisis in T2 deficiency, often with basal ganglia lesions. However, we should reconsider neurological problems in T2 deficiency. Some patients developed neurological manifestations that did not associate with apparent severe metabolic crises [9, 13, 31, 32, 35, 38, 46, 47, 52, 68]. Here, we focus on neurological manifestation that is not associated with severe metabolic crises.
In 1994, Ozand et al. [52] reported four T2-deficient patients with delayed development before their first acidotic events. Basal ganglia lesions were identified in three of them by MRI imaging after the metabolic episodes. Ozand et al. first reviewed the neurological problem in T2 deficiency and summarized eight patients from previous case reports who had neurological symptoms. Among them, one patient (GK02) also had delayed development in infancy before the first crisis [68].
Before 2013, although neurological manifestation before a first metabolic crisis was presented in some T2-deficient patients (GK06 [9] GK12 [13], GK32 [32], and GK41 [31]), most such patients presented with motor delay and hypotonia, and extrapyramidal manifestation became evident after their metabolic crisis, as shown in Table 4. Little attention was paid to extrapyramidal manifestation before or without severe metabolic crisis. In 2013, we described a peculiar case, GK84, who suffered hypotonia since 4 months of age and continuous involuntary upper extremity movements since 10 months of age [38]. His development was delayed, and at 4 years of age daily activities were difficult because of involuntary movements and MRI showed bilateral T2 hyperintensities in both the putamen and cerebral peduncles. His first metabolic crisis occurred at 5 years of age following gastroenteritis. This case clearly showed that extrapyramidal abnormalities can occur without apparent metabolic crisis in some T2-deficient patients. Retrospectively, extrapyramidal manifestation was also seen in GK36 and GK41. Hypotonia and developmental delay in other patients were also possibly caused by extrapyramidal abnormality. A recent report by Paquay et al. [47] supports our finding. They focused on neurological involvements in the series of 26 French T2-deficient patients. Among them, two patients who had never had an apparent ketoacidotic crisis, showed neurological symptoms, including motor delay, hypotonia, ataxia, and dystonia since infancy or early childhood. Another two patients had motor delay, hypotonia, dyskinesia before their first crisis and another patient developed choreoathetosis and dystonia from the age of 15 years after a long interval from her first and only ketoacidotic crisis. Basal ganglia lesions were identified in all these cases.
Neurological manifestations and basal ganglia lesions are apparently associated with severe metabolic crises in T2 deficiency. However, based on the above findings, neurological manifestations, mainly extrapyramidal manifestations, may occur without severe metabolic decompensation in infancy or early childhood and may be worsened after severe metabolic crises in some T2-deficient patients. It should be stressed that a large number of T2-deficient patients do not develop neurological manifestation even after metabolic crisis.
The pathogenesis of neurological manifestation and basal ganglia lesions remains unclear. In propionic acidemia and methylmalonic acidemia, basal ganglia lesions are also regarded as sequela of severe metabolic decompensation [69,70,71,72]. However, extrapyramidal manifestation and basal ganglia lesions without severe metabolic episodes have also been described in these disorders [73, 74]. In HSD10 disease, there is a defect of the step just upstream from T2 in the isoleucine-catabolic pathway and basal ganglia lesions have also been reported [75, 76]. However, patients with SCOT deficiency, another ketolytic defect, also developed severe ketoacidosis but no basal ganglia lesions have been reported in SCOT deficiency. SCOT deficiency affects only ketolysis and T2 deficiency affects both ketolysis and isoleucine catabolism. Hence, it is reasonable that neurological manifestation is attributed to impairment of isoleucine catabolism.
Isoleucine is catabolized within the rat brain and serves as fuel for brain energy metabolism [77]. Hence, blockage of the isoleucine-catabolic pathway may result in accumulation of tiglyl-CoA, 2M3HB-CoA, and 2-methylacetoacetyl-CoA and Coenzyme A sequestration, toxicity or redistribution (CASTOR), as proposed by Mitchell et al. [78]. Does accumulation of 2M3HB and 2AcAc result in basal ganglia lesions, leading to extrapyramidal manifestation? There are only a few reports about a direct influence of these compounds on brain function [79, 80]. They showed that these compounds inhibited aerobic energy metabolism in the TCA cycle and the mitochondrial respiratory chain and induced oxidative stress in rat brain cortex in vitro. The basal ganglia may be vulnerable if these compounds affect the human brain in vivo in a similar way.
However, these findings cannot explain why some patients have such basal ganglia lesions without severe metabolic crises. At present, we cannot predict whether a T2-deficient patient will have neurological manifestation or not. Genotype does not appear to be associated with clinical manifestation including neurological manifestation. It is possible that other genetic or environmental factors, which may enhance the toxic effect of isoleucine metabolites, are associated with basal ganglia lesions. One possible environmental factor is excess isoleucine load. Mild protein restriction (1.5–2.0 g/kg/day) has been recommended from a theoretical standpoint but is not evidence-based [5, 65]; there is no data whether this level of protein restriction results in reduced 2M3HB and 2MAcAc levels or reduced intra-brain isoleucine load. One possible genetic factor is the efficiency of monocarboxylate transporters that function through the mitochondrial and cell membrane. 2M3HB and 2AcAc are monocarboxylates; therefore, such transporters may determine intracellular or intramitochondrial concentration of 2M3HB and 2MAcAc.
Lastly, how to manage T2-deficient babies who are asymptomatic and identified by NBS or familial analysis? Fasting avoidance and sufficient glucose supplementation (including intravenous infusion) under ketogenic stresses are the basis of long-term management. Should we apply mild protein restriction to all T2-deficient newborns? Theoretically, isoleucine overload should be avoided. There is, however, no evidence for effectiveness of this treatment. Paquay et al. compared six patients with neurological manifestation and 20 patients without it and could not find any relationship between protein intake and neurological manifestation [47]. To reduce intramitochondrial accumulation of isoleucine metabolites, including their CoA-conjugates, L-Carnitine supplementation is also theoretically reasonable. However, L-Carnitine supplementation does not seem to be effective for prevention of neurological manifestation because L-Carnitine was supplemented in all of the six patients with neurological manifestation in Paquay’s series [47]. Because this disorder is so rare, it is difficult to perform a prospective clinical trial to study whether protein restriction and L-Carnitine supplementation are protective against neurological manifestation. We also need to understand metabolic pathophysiology in the brain to elucidate the pathogenesis of neurological manifestation.
References
Daum RS, Lamm PH, Mamer OA, Scriver CRA. “new” disorder of isoleucine catabolism. Lancet. 1971;2:1289–90.
Daum RS, Scriver CR, Mamer OA, Delvin E, Lamm P, Goldman H. An inherited disorder of isoleucine catabolism causing accumulation of alpha-methylacetoacetate and alpha-methyl-beta -hydroxybutyrate, and intermittent metabolic acidosis. Pediatr Res. 1973;7:149–60.
Fukao T, Maruyama S, Ohura T, Hasegawa Y, Toyoshima M, Haapalainen AM, et al. Three Japanese patients with beta-ketothiolase deficiency who share a mutation, c.431A>C (H144P) in ACAT1: subtle abnormality in urinary organic acid analysis and blood acylcarnitine analysis using tandem mass spectrometry. JIMD Rep. 2012;3:107–15.
Yamaguchi S, Orii T, Sakura N, Miyazawa S, Hashimoto T. Defect in biosynthesis of mitochondrial acetoacetyl-coenzyme A thiolase in cultured fibroblasts from a boy with 3-ketothiolase deficiency. J Clin Invest. 1988;81:813–7.
Mitchell GA, Fukao T. Inborn errors of ketone body metabolism. In: Scriver CR, Beaudet AL, Sly WS & Valle D, editors. The metabolic & molecular basis of inherited disease. Vol. 2, Ch. 102 New York: McGraw-Hill; 2001. p. 2327–56.
Fukao T, Mitchell G, Sass JO, Hori T, Orii K, Aoyama Y. Ketone body metabolism and its defects. J Inherit Metab Dis. 2014;37:541–51.
Abdelkreem E, Otsuka H, Sasai H, Aoyama Y, Hori T, Abd El Aal M, et al. Beta-ketothiolase deficiency: resolving challenges in diagnosis. J Inborn Errors Metab Screen. 2016;4:1–9.
Fukao T, Yamaguchi S, Nagasawa H, Kano M, Orii T, Fujiki Y, et al. Molecular cloning of cDNA for human mitochondrial acetoacetyl-CoA thiolase and molecular analysis of 3-ketothiolase deficiency. J Inherit Metab Dis. 1990;13:757–60.
Fukao T, Yamaguchi S, Tomatsu S, Orii T, Frauendienst-Egger G, Schrod L, et al. Evidence for a structural mutation (347Ala to Thr) in a German family with 3-ketothiolase deficiency. Biochem Biophys Res Commun. 1991;179:124–9.
Kano M, Fukao T, Yamaguchi S, Orii T, Osumi T, Hashimoto T. Structure and expression of the human mitochondrial acetoacetyl-CoA thiolase-encoding gene. Gene. 1991;109:285–90.
Fukao T, Yamaguchi S, Orii T, Osumi T, Hashimoto T. Molecular basis of 3-ketothiolase deficiency: identification of an AG to AC substitution at the splice acceptor site of intron 10 causing exon 11 skipping. Biochim Biophys Acta. 1992;1139:184–8.
Fukao T, Yamaguchi S, Wakazono A, Okamoto H, Orii T, Osumi T, et al. Molecular basis of 3-ketothiolase deficiency: detection of gene mutations and expression of mutant cDNAs of mitochondrial acetoacetyl-CoA thiolase. J Inherit Metab Dis. 1992;15:815–20.
Wajner M, Sanseverino MT, Giugliani R, Sweetman L, Yamaguchi S, Fukao T, et al. Biochemical investigation of a Brazilian patient with a defect in mitochondrial acetoacetylcoenzyme-A thiolase. Clin Genet. 1992;41:202–5.
Masuno M, Kano M, Fukao T, Yamaguchi S, Osumi T, Hashimoto T, et al. Chromosome mapping of the human mitochondrial acetoacetyl-coenzyme A thiolase gene to 11q22.3----q23.1 by fluorescence in situ hybridization. Cytogenet Cell Genet. 1992;60:121–2.
Fukao T, Yamaguchi S, Orii T, Schutgens RB, Osumi T, Hashimoto T. Identification of three mutant alleles of the gene for mitochondrial acetoacetyl-coenzyme A thiolase. A complete analysis of two generations of a family with 3-ketothiolase deficiency. J Clin Invest. 1992;89:474–9.
Fukao T, Yamaguchi S, Scriver CR, Dunbar G, Wakazono A, Kano M, et al. Molecular studies of mitochondrial acetoacetyl-coenzyme A thiolase deficiency in the two original families. Hum Mutat. 1993;2:214–20.
Fukao T, Yamaguchi S, Wakazono A, Orii T, Hoganson G, Hashimoto T. Identification of a novel exonic mutation at -13 from 5’ splice site causing exon skipping in a girl with mitochondrial acetoacetyl-coenzyme A thiolase deficiency. J Clin Invest. 1994;93:1035–41.
Fukao T, Song XQ, Yamaguchi S, Orii T, Wanders RJ, Poll-The BT, et al. Mitochondrial acetoacetyl-coenzyme A thiolase gene: a novel 68-bp deletion involving 3’ splice site of intron 7, causing exon 8 skipping in a Caucasian patient with beta-ketothiolase deficiency. Hum Mutat. 1995;5:94–96.
Fukao T, Kodama A, Aoyanagi N, Tsukino R, Uemura S, Song XQ, et al. Mild form of beta-ketothiolase deficiency (mitochondrial acetoacetyl-CoA thiolase deficiency) in two Japanese siblings: identification of detectable residual activity and cross-reactive material in EB-transformed lymphocytes. Clin Genet. 1996;50:263–6.
Fukao T, Song XQ, Yamaguchi S, Kondo N, Orii T, Matthieu JM, et al. Identification of three novel frameshift mutations (83delAT, 754insCT, and 435+1G to A) of mitochondrial acetoacetyl-coenzyme A thiolase gene in two Swiss patients with CRM-negative beta-ketothiolase deficiency. Hum Mutat. 1997;9:277–9.
Wakazono A, Fukao T, Yamaguchi S, Hori T, Orii T, Lambert M, et al. Molecular, biochemical, and clinical characterization of mitochondrial acetoacetyl-coenzyme A thiolase deficiency in two further patients. Hum Mutat. 1995;5:34–42.
Fukao T, Nakamura H, Song XQ, Nakamura K, Orii KE, Kohno Y, et al. Characterization of N93S, I312T, and A333P missense mutations in two Japanese families with mitochondrial acetoacetyl-CoA thiolase deficiency. Hum Mutat. 1998;12:245–54.
Sewell AC, Herwig J, Wiegratz I, Lehnert W, Niederhoff H, Song XQ, et al. Mitochondrial acetoacetyl-CoA thiolase (beta-ketothiolase) deficiency and pregnancy. J Inherit Metab Dis. 1998;21:441–2.
Nakamura K, Fukao T, Perez-Cerda C, Luque C, Song XQ, Naiki Y, et al. A novel single-base substitution (380C>T) that activates a 5-base downstream cryptic splice-acceptor site within exon 5 in almost all transcripts in the human mitochondrial acetoacetyl-CoA thiolase gene. Mol Genet Metab. 2001;72:115–21.
Fukao T, Scriver CR, Kondo N. The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab. 2001;72:109–14.
Fukao T, Nakamura H, Nakamura K, Perez-Cerda C, Baldellou A, Barrionuevo CR, et al. Characterization of six mutations in five Spanish patients with mitochondrial acetoacetyl-CoA thiolase deficiency: effects of amino acid substitutions on tertiary structure. Mol Genet Metab. 2002;75:235–43.
Fukao T, Matsuo N, Zhang GX, Urasawa R, Kubo T, Kohno Y, et al. Single base substitutions at the initiator codon in the mitochondrial acetoacetyl-CoA thiolase (ACAT1/T2) gene result in production of varying amounts of wild-type T2 polypeptide. Hum Mutat. 2003;21:587–92.
Fukao T, Zhang GX, Sakura N, Kubo T, Yamaga H, Hazama A, et al. The mitochondrial acetoacetyl-CoA thiolase (T2) deficiency in Japanese patients: urinary organic acid and blood acylcarnitine profiles under stable conditions have subtle abnormalities in T2-deficient patients with some residual T2 activity. J Inherit Metab Dis. 2003;26:423–31.
Zhang GX, Fukao T, Rolland MO, Zabot MT, Renom G, Touma E, et al. Mitochondrial acetoacetyl-CoA thiolase (T2) deficiency: T2-deficient patients with “mild” mutation(s) were previously misinterpreted as normal by the coupled assay with tiglyl-CoA. Pediatr Res. 2004;56:60–64.
Mrazova L, Fukao T, Halovd K, Gregova E, Kohut V, Pribyl D, et al. Two novel mutations in mitochondrial acetoacetyl-CoA thiolase deficiency. J Inherit Metab Dis. 2005;28:235–6.
Zhang G, Fukao T, Sakurai S, Yamada K, Michael Gibson K, Kondo N. Identification of Alu-mediated, large deletion-spanning exons 2-4 in a patient with mitochondrial acetoacetyl-CoA thiolase deficiency. Mol Genet Metab. 2006;89:222–6.
Sakurai S, Fukao T, Haapalainen AM, Zhang G, Yamada K, Lilliu F, et al. Kinetic and expression analyses of seven novel mutations in mitochondrial acetoacetyl-CoA thiolase (T2): identification of a Km mutant and an analysis of the mutational sites in the structure. Mol Genet Metab. 2007;90:370–8.
Fukao T, Boneh A, Aoki Y, Kondo N. A novel single-base substitution (c.1124A>G) that activates a 5-base upstream cryptic splice donor site within exon 11 in the human mitochondrial acetoacetyl-CoA thiolase gene. Mol Genet Metab. 2008;94:417–21.
Fukao T, Horikawa R, Naiki Y, Tanaka T, Takayanagi M, Yamaguchi S, et al. A novel mutation (c.951C>T) in an exonic splicing enhancer results in exon 10 skipping in the human mitochondrial acetoacetyl-CoA thiolase gene. Mol Genet Metab. 2010;100:339–44.
Fukao T, Nguyen HT, Nguyen NT, Vu DC, Can NT, Pham AT, et al. A common mutation, R208X, identified in Vietnamese patients with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet Metab. 2010;100:37–41.
Thummler S, Dupont D, Acquaviva C, Fukao T, de Ricaud D. Different clinical presentation in siblings with mitochondrial acetoacetyl-CoA thiolase deficiency and identification of two novel mutations. Tohoku J Exp Med. 2010;220:27–31.
Fukao T, Aoyama Y, Murase K, Hori T, Harijan RK, Wierenga RK, et al. Development of MLPA for human ACAT1 gene and identification of a heterozygous Alu-mediated deletion of exons 3 and 4 in a patient with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet Metab. 2013;110:184–7.
Buhas D, Bernard G, Fukao T, Decarie JC, Chouinard S, Mitchell GA. A treatable new cause of chorea: beta-ketothiolase deficiency. Mov Disord. 2013;28:1054–6.
Akella RR, Aoyama Y, Mori C, Lingappa L, Cariappa R, Fukao T. Metabolic encephalopathy in beta-ketothiolase deficiency: the first report from India. Brain Dev. 2014;36:537–40.
Otsuka H, Sasai H, Nakama M, Aoyama Y, Abdelkreem E, Ohnishi H, et al. Exon 10 skipping in ACAT1 caused by a novel c. 949G>A mutation located at an exonic splice enhancer site. Mol Med Rep. 2016;14:4906–10.
Nguyen KN, Abdelkreem E, Colombo R, Hasegawa Y, Can NTB, Bui TP, et al. Characterization and outcome of 41 patients with beta-ketothiolase deficiency: 10 years’ experience of a medical center in northern Vietnam. J Inherit Metab Dis. 2017;40:395–401.
Abdelkreem E, Akella RRD, Dave U, Sane S, Otsuka H, Sasai H. et al. Clinical and mutational characterizations of ten Indian patients with beta-ketothiolase deficiency. JIMD Rep. 2017;35:59–65.
Sasai H, Aoyama Y, Otsuka H, Abdelkreem E, Nakama M, Hori T, et al. Single-nucleotide substitution T to A in the polypyrimidine stretch at the splice acceptor site of intron 9 causes exon 10 skipping in the ACAT1 gene. Mol Genet Genom Med. 2017;5:177–84.
Abdelkreem E, Alobaidy H, Aoyama Y, Mahmoud S, Abd El Aal M, Fukao T. Two Libyan siblings with beta-ketothiolase deficiency: a case report and review of literature. Egypt J Med Human Genet. 2017;18:199–203.
Aoyama Y, Sasai H, Abdelkreem E, Otsuka H, Nakama M, Kumar S, et al. A novel mutation (c.12113T>A) in the polypyrimidine tract of the splice acceptor site of intron 2 causes exon 3 skipping in mitochondrial acetoacetyl-CoA thiolase gene. Mol Med Rep. 2017;15:3879–84.
Grunert SC, Schmitt RN, Schlatter SM, Gemperle-Britschgi C, Balci MC, Berg V, et al. Clinical presentation and outcome in a series of 32 patients with 2-methylacetoacetyl-coenzyme A thiolase (MAT) deficiency. Mol Genet Metab. 2017;122:67–75.
Paquay S, Bourillon A, Pichard S, Benoist JF, de Lonlay P, Dobbelaere D, et al. Mitochondrial acetoacetyl-CoA thiolase deficiency: basal ganglia impairment may occur independently of ketoacidosis. J Inherit Metab Dis. 2017;40:415–22.
Sarafoglou K, Matern D, Redlinger-Grosse K, Bentler K, Gaviglio A, Harding CO, et al. Siblings with mitochondrial acetoacetyl-CoA thiolase deficiency not identified by newborn screening. Pediatrics. 2011;128:e246–50.
Wojcik MH, Wierenga KJ, Rodan LH, Sahai I, Ferdinandusse S, Genetti CA, et al. Beta-ketothiolase deficiency presenting with metabolic stroke after a normal newborn screen in two individuals. JIMD Rep. 2017;39:45–54.
Estrella J, Wilcken B, Carpenter K, Bhattacharya K, Tchan M, Wiley V. Expanded newborn screening in New South Wales: missed cases. J Inherit Metab Dis. 2014;37:881–7.
Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil SD, Chaing SH, et al. The tandem mass spectrometry newborn screening experience in North Carolina: 1997-2005. J Inherit Metab Dis. 2006;29:76–85.
Ozand PT, Rashed M, Gascon GG, al Odaib A, Shums A, Nester M, et al. 3-Ketothiolase deficiency: a review and four new patients with neurologic symptoms. Brain Dev. 1994;16:38–45.
Middleton B. The oxoacyl-coenzyme A thiolases of animal tissues. Biochem J. 1973;132:717–30.
Middleton B, Bartlett K. The synthesis and characterisation of 2-methylacetoacetyl coenzyme A and its use in the identification of the site of the defect in 2-methylacetoacetic and 2-methyl-3-hydroxybutyric aciduria. Clin Chim Acta. 1983;128:291–305.
Erdol S, Ture M, Yakut T, Saglam H, Sasai H, Abdelkreem E, et al. A Turkish patient with succinyl-CoA: 3-oxoacid CoA transferase deficiency mimicking diabetic ketoacidosis. J Inborn Errors Metab Screen. 2016;4:1–5.
Attia N, Sakati N, al Ashwal A, al Saif R, Rashed M, Ozand PT. Isovaleric acidemia appearing as diabetic ketoacidosis. J Inherit Metab Dis. 1996;19:85–86.
Dweikat IM, Naser EN, Abu Libdeh AI, Naser OJ, Abu Gharbieh NN, Maraqa NF, et al. Propionic acidemia mimicking diabetic ketoacidosis. Brain Dev. 2011;33:428–31.
Guven A, Cebeci N, Dursun A, Aktekin E, Baumgartner M, Fowler B. Methylmalonic acidemia mimicking diabetic ketoacidosis in an infant. Pediatr Diabetes. 2012;13:e22–25.
Bonnefont JP, Specola NB, Vassault A, Lombes A, Ogier H, de Klerk JB, et al. The fasting test in paediatrics: application to the diagnosis of pathological hypo- and hyperketotic states. Eur J Pediatr. 1990;150:80–85.
Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis. 2012;35:81–89.
Zschocke J, Ruiter JP, Brand J, Lindner M, Hoffmann GF, Wanders RJ, et al. Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr Res. 2000;48:852–5.
Robinson BH, Sherwood WG, Taylor J, Balfe JW, Mamer OA. Acetoacetyl CoA thiolase deficiency: a cause of severe ketoacidosis in infancy simulating salicylism. J Pediatr. 1979;95:228–33.
Fukao T, Song XQ, Mitchell GA, Yamaguchi S, Sukegawa K, Orii T, et al. Enzymes of ketone body utilization in human tissues: protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr Res. 1997;42:498–502.
Gibson KM, Lee CF, Kamali V, Sovik O. A coupled assay detecting defects in fibroblast isoleucine degradation distal to enoyl-CoA hydratase: application to 3-oxothiolase deficiency. Clin Chim Acta. 1992;205:127–35.
Hori T, Yamaguchi S, Shinkaku H, Horikawa R, Shigematsu Y, Takayanagi M, et al. Inborn errors of ketone body utilization. Pediatr Int. 2015;57:41–48.
Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: selective screening vs. expanded newborn screening. Mol Genet Metab Rep. 2018;16:5–10.
Akagawa S, Fukao T, Akagawa Y, Sasai H, Kohdera U, Kino M, et al. Japanese male siblings with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) without neurological regression. JIMD Rep. 2017;32:81–85.
Middleton B, Bartlett K, Romanos A, Gomez Vazquez J, Conde C, Cannon RA, et al. 3-Ketothiolase deficiency. Eur J Pediatr. 1986;144:586–9.
Haas RH, Marsden DL, Capistrano-Estrada S, Hamilton R, Grafe MR, Wong W, et al. Acute basal ganglia infarction in propionic acidemia. J Child Neurol. 1995;10:18–22.
Hamilton RL, Haas RH, Nyhan WL, Powell HC, Grafe MR. Neuropathology of propionic acidemia: a report of two patients with basal ganglia lesions. J Child Neurol. 1995;10:25–30.
Heidenreich R, Natowicz M, Hainline BE, Berman P, Kelley RI, Hillman RE, et al. Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. J Pediatr. 1988;113:1022–7.
Thompson GN, Christodoulou J, Danks DM. Metabolic stroke in methylmalonic acidemia. J Pediatr. 1989;115:499–500.
Prada CE, Villamizar-Schiller IT. Globus pallidus involvement as initial presentation of methylmalonic acidemia. Mov Disord. 2014;29:870.
Scholl-Burgi S, Haberlandt E, Gotwald T, Albrecht U, Baumgartner Sigl S, Rauchenzauner M, et al. Stroke-like episodes in propionic acidemia caused by central focal metabolic decompensation. Neuropediatrics. 2009;40:76–81.
Cazorla MR, Verdu A, Perez-Cerda C, Ribes A. Neuroimage findings in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency. Pediatr Neurol. 2007;36:264–7.
Sass JO, Forstner R, Sperl W. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: impaired catabolism of isoleucine presenting as neurodegenerative disease. Brain Dev. 2004;26:12–4.
Murin R, Mohammadi G, Leibfritz D, Hamprecht B. Glial metabolism of isoleucine. Neurochem Res. 2009;34:194–204.
Mitchell GA, Gauthier N, Lesimple A, Wang SP, Mamer O, Qureshi I. Hereditary and acquired diseases of acyl-coenzyme A metabolism. Mol Genet Metab. 2008;94:4–15.
Rosa RB, Schuck PF, de Assis DR, Latini A, Dalcin KB, Ribeiro CA, et al. Inhibition of energy metabolism by 2-methylacetoacetate and 2-methyl-3-hydroxybutyrate in cerebral cortex of developing rats. J Inherit Metab Dis. 2005;28:501–15.
Leipnitz G, Seminotti B, Amaral AU, Fernandes CG, Dutra-Filho CS, Wajner M. Evidence that 2-methylacetoacetate induces oxidative stress in rat brain. Metab Brain Dis. 2010;25:261–7.
Acknowledgements
We thank Professor emeritus Tadao Orii (Gifu University) and Professor emeritus Seiji Yamaguchi (Shimane University) for their mentorship, Professor Grant Mitchell and Professor Oliver Sass for long-term collaborations and for discussions on defective ketone body metabolism. We also thank Ms Naomi Sakaguchi for her dedicated assistance with laboratory work. This research was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan [Grant Numbers 16K09962, 15K01693], by AMED under Grant Number JP17ek0109276, and by Health and Labour Sciences Research Grants (H29-nanchitou(nan)-ippan-051) for Research on rare and intractable diseases. We thank Jeremy Allen, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
This research was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan [Grant Numbers 16K09962, 15K01693], by AMED under Grant Number JP17ek0109276, and by Health and Labour Sciences Research Grants [H29-nanchitou(nan)-ippan-051] for Research on rare and intractable diseases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Fukao, T., Sasai, H., Aoyama, Y. et al. Recent advances in understanding beta-ketothiolase (mitochondrial acetoacetyl-CoA thiolase, T2) deficiency. J Hum Genet 64, 99–111 (2019). https://doi.org/10.1038/s10038-018-0524-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0524-x
This article is cited by
-
Hepatic Acat2 overexpression promotes systemic cholesterol metabolism and adipose lipid metabolism in mice
Diabetologia (2023)
-
C4OH is a potential newborn screening marker—a multicenter retrospective study of patients with beta-ketothiolase deficiency in China
Orphanet Journal of Rare Diseases (2021)
-
2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency: one disease - two pathways
Orphanet Journal of Rare Diseases (2020)
