
Abstract
RAD51D gene’s protein product is known to be involved in the DNA repair mechanism by homologous recombination. RAD51D germline mutations have been recently associated with ovarian and breast cancer (OC and BC, respectively) predisposition. Our aim was to evaluate the frequency of hereditary RAD51D mutations in Greek patients. To address this, we have screened for RAD51D germline mutations 609 BRCA1- and BRCA2-negative patients diagnosed with OC, unselected for age or family history, and 569 BC patients diagnosed under 55 years and with an additional relative with BC or OC. We identified four pathogenic mutations in four unrelated individuals with family history of BC and/or OC. Three of the RAD51D carriers had developed BC, while the other one was an OC patient, thus accounting for a mutation frequency of 0.16% in the OC cohort and 0.53% in the BC cohort. One of the detected mutations is novel (c.738 + 1G > A), whereas the rest had been detected previously (p.Gln151Ter, p.Arg186Ter, and p.Arg300Ter). It is noteworthy that the 4 carrier families had 13 BC cases and only 4 OC cases. Our data support that RAD51D should be implemented into the comprehensive multigene panel, as mutation carriers may benefit from the administration of PARP inhibitors.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the most commonly diagnosed cancer among women [1], while ovarian cancer (ΟC) still remains the gynecological malignancy with the highest mortality [2]. BRCA1/2 were the first genes associated with hereditary BC and OC [3, 4]. Recently, several genes, the protein products of which are involved in homologous recombination and Fanconi anemia pathways, were implicated in BC/OC predisposition [5, 6].
Of these, RAD51D gene is rather interesting since its impaired function is clearly associated with elevated OC risk, but its implication in BC predisposition is rather controversial. RAD51D is a member of the RAD51 protein family and a constituent of DNA repair mechanism by homologous recombination through the BCDX2 complex formation, which binds to single-stranded DNA after damage and provides homology detection between the damaged and wild-type strand in the repair process [7, 8].
RAD51D was analyzed sequentially in families with multiple cases of BC and/or OC, after the first report of association of RAD51C mutations with BC/OC susceptibility, where in a cohort of 911 probands and 1060 controls, 0.88% and 0.09% carried loss-of-function (LoF) RAD51D alleles, respectively. A prominent prevalence of LoF alleles was observed in families with elevated OC burden while, interestingly, no association was observed with BC predisposition alone [9]. In subsequent studies, the association of LoF RAD51D mutations to OC predisposition was replicated and more specifically, they were detected in approximately 1–2% of the OC patients tested, depending on the OC burden in the family relatives. In line with the initial study conducted, several following reports failed to demonstrate an association between RAD51D deleterious mutations and BC predisposition [2, 10, 11]. Therefore, RAD51D has been considered as a moderate penetrance OC susceptibility gene, conferring a 10–12% lifetime risk, when mutated [12], whereas its association with BC predisposition remained unclear. However, very recently there has been an accumulation of data addressing the latter, still unresolved association.
Couch et al. [13] identified pathogenic RAD51D mutations in triple-negative BC patients unselected for family history, suggesting an association with this disease subtype, whereas subsequent large-scale studies performing panel testing followed. Specifically, ~ 35,000 BC patients were tested using a 25-gene panel, where the RAD51D mutation prevalence was 0.6%; in a sub-cohort of 692 carriers diagnosed with triple-negative BC, the mutation prevalence rose up to 0.9% [14]. In addition, in a US nationwide sample group of women diagnosed with BC, the presence of RAD51D mutations, although exhibiting a low frequency (0.07%), was significantly associated with a moderate BC risk (odds ratio (OR), 3.07; 95% confidence interval (CI) 1.21–7.88) [15].
On these grounds, we can argue that pathogenic germline mutations in RAD51D probably contribute to predisposition in both BC and OC, whereas the specific association of such mutations to the development of BC or OC, in terms of penetrance and severity, is less clear. On the molecular level, when the role of the encoded protein in the biochemical pathway of DNA repair by homologous recombination is taken into account, it would not be unexpected for pathogenic mutations to be associated with the development of both forms of familial cancer.
The present study is an attempt to define the prevalence of RAD51D mutations in a large cohort of Greek OC and BC patients, tested negative for mutations in BRCA1 and BRCA2, hoping to elucidate the contribution of this gene to the so-called missing heritability and to further shed light on its association with BC and OC.
Materials and methods
Study population
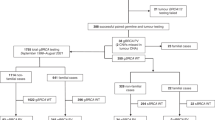
A total of 609 patients diagnosed with epithelial OC, unselected for age or family history, were recruited ad hoc from various Greek hospitals between 2006 and 2016. Mean age at diagnosis was 55 years, ranging from 19 to 87 years old. Subsequently, 569 BC patients were included in the study, fulfilling the following criteria: (a) diagnosis of invasive BC at or below 55 years of age and (b) at least one first- or second-degree relative diagnosed with BC or OC. BC patients were also recruited ad hoc from various Greek hospitals between 2006 and 2015. Mean age at diagnosis was 42 years, ranging from 20 to 55 years old. All patients included in the present study have been previously tested negative for germline mutations in the BRCA1 and BRCA2 genes. The study was approved by the Bioethics Committee of NCSR “Demokritos” (240/ΕΗΔ/11.3) and was in agreement with the 1975 Helsinki Statement, revised in 1983. Written informed consent was obtained from all study participants.
DNA extraction
Total genomic DNA was isolated from peripheral blood lymphocytes following the salt extraction procedure proposed by Miller et al. [16].
Germline mutation screening
The entire coding region of RAD51D was PCR amplified and sequenced on an ABI 3130XL Genetic Analyzer (ThermoFisher Scientific, Carlsbad, CA, USA). All variants were named according to Human Genome Variation Society guidelines, whereas NM_002878.3 was used as the reference sequence [17]. Primer sequences and protocols are available upon request.
In silico prediction tools
A variety of in silico tools and simulation protein model resources were used for the assessment of the variants of unknown clinical significance (VUS). Align GVGD (Grantham Variation, Grantham Deviation) [18, 19] and MutationTaster [20] were used to classify the effect of missense variants on the protein formation, whereas PhastCons [21] was used to assess evolutionary conservation. Minor allele frequencies for the European population were retrieved from Exome Aggregation Consortium (ExAC) [22]. Several splicing prediction tools were used to assess the impact of intronic variants (Human Splicing finder (http://www.umd.be/HSF3/) [23], ESE finder [24], MaxEntScan [25], Netgene2 [26], SplicePort [27], and NNSplice [28]). The Swiss-Model server (https://swissmodel.expasy.org/) was employed to construct a homology model of RAD51D, as no experimentally derived three-dimensional structure of full-length RAD51D [29] was available at the time of writing this manuscript.
RNA extraction and RT-PCR
To evaluate the possible splicing effect of the c.738 + 1 G > A variant, total RNA was extracted from peripheral blood lymphocytes using Trizol (ThermoFisher Scientific) following manufacturer’s instructions. Subsequently, cDNA was amplified to generate the wild-type and mutant isoforms.
Comparison of cases and controls
For each of the two cohorts (BC and OC), frequencies of RAD51D pathogenic/likely pathogenic variants were compared with frequencies of RAD51D pathogenic variants in the ExAC non-Finnish European population reference control dataset, excluding data deriving from The Cancer Genome Atlas exomes [22, 30]. In addition, 76 cancer-free individuals of Greek descent were sequenced for the entire coding sequence of RAD51D.
Multigene panel testing of RAD51D-positive samples by next-generation sequencing
Samples tested positive for a RAD51D germline mutation were subsequently subjected to multigene panel testing to exclude the possibility of the presence of another causative mutation. Germline DNA was used to prepare indexed libraries to target the sequence of 94-cancer predisposing genes using the Illumina Trusight Cancer Panel and were sequenced on a MiSeq analyzer (Illumina, San Diego, USA). FASTQ, BAM, and VCF files were produced through Basespace (Illumina). The minimum base and amplicon coverage was 50× and 100× , respectively. All called variants of interest were confirmed by Sanger sequencing. Called variants were annotated by VariantStudio version 3 (Illumina) against the human reference genome GRCh38.
Results
RAD51D pathogenic mutations in OC patients
We have screened 609 Greek epithelial OC patients, unselected for age or family history, for the presence of RAD51D germline mutations. All patients have been previously tested negative for BRCA1/2 mutations. Of those, 124 (20.4%) had reported family history, defined as having at least one first- or second-degree relative presented with BC at or below the age of 50 years and/or one relative with OC. Tumor histology was mainly serous (73%), of which about 60% corresponding to high grade (Supplementary Table 1).
Screening revealed a novel nucleotide change, c.738 + 1 G > Α, affecting the acceptor site of exon 8, demanding further investigation, which was detected in a patient diagnosed with high-grade serous OC at the age of 65 years. Her sister was also diagnosed with OC (Fig.1, family 510). cDNA analysis revealed aberrant splicing, with partial retention of 37 intronic bases (Fig. 2), causing a frameshift and a premature termination codon (p.Val246fs*92) and classified as pathogenic, according to ACMG guidelines (PVS1 rule, very strong evidence) [31, 32].

Abridged pedigrees of four families with RAD51D mutations. Individuals with breast cancer are shown as black circles. Individuals with ovarian cancer are shown as pink circles. Other cancers are presented with other colors, explained within the pedigree. Probands are indicated by an arrow. BC breast cancer, Ca cancer, CRC colorectal cancer, OvCa ovarian cancer

RNA analysis of the RAD51D c.738+1 G > A pathogenic mutation. a Sanger sequencing of genomic DNA from patient 510 showing the G > A substitution in heterozygosity in the first intronic base. b Agarose gel electropherogram of cDNA products from patient 510 compared with control. c Sanger sequencing of cDNA from patient 510 showing the intron retention. d Illustration of intron retention
Therefore, the frequency of pathogenic RAD51D mutations in our OC cohort is 0.16% (1/609), overall. RAD51D pathogenic mutations were statistically more frequent, but not significant, among OC cases when compared to controls from the ExAC dataset (OR: 8.79; 95% CI: 0.19–78.73; p = 0.1264). Our analysis did not reach statistical significance due to the small number of carriers, i.e., one case, detected in our series. RAD51D sequencing among Greek cancer-free individuals did not reveal any pathogenic/likely pathogenic variants.
RAD51D pathogenic mutations in BC patients
BC patients previously tested negative for BRCA1/2 mutations were also screened for RAD51D mutations. Selection criteria aimed to define a higher-risk group with a greater probability of hereditary etiology and were therefore selected as described above. Tumor pathology characteristics of the 569 BC patients, fulfilling our selection criteria are shown in Supplementary Table 2.
Three patients of our cohort carried definitely or likely pathogenic variants, accounting for a mutation frequency of 0.53% (3/569) in this BC patient group. RAD51D pathogenic variants were statistically significant, more frequent among BC cases, when compared with data deriving from the ExAC dataset (p = 0.0005). Moreover, RAD51D sequencing among Greek cancer-free individuals did not reveal any pathogenic/likely pathogenic variants.
A 33-year-old patient, diagnosed with a luminal B, ductal invasive BC, having two paternal aunts diagnosed with BC > 50 years, was found to carry the c.898 C > T (p.Arg300Ter) mutation (family 487). The truncating variant, located on exon 9, has already been characterized as likely pathogenic in ClinVar [13, 33].
The c.451 C > T (p.Gln151Ter) mutation, located in exon 5, was detected in a 55-year-old woman (family 752) diagnosed with luminal A ductal invasive BC and a very strong family history, including a sister who was diagnosed with both BC and OC, and four more BC diagnoses among relatives, two of which being < 35 years. The c.556 C > T (p.Arg186Ter) mutation, located in exon 6, was detected in a 43-year-old, triple-negative BC patient (family 2247), who had two maternal aunts, both diagnosed with BC at age 50 years. The mean age of BC diagnosis among RAD51D carriers was 43.6 years. Detailed pedigrees of all carriers are depicted in Fig.1.
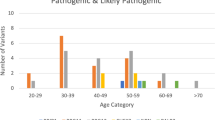
All pathogenic/likely pathogenic RAD51D variants identified during our study, along with patients’ tumor characteristics and family history, are listed in Table 1.
Further screening of RAD51D mutation carriers for causative mutations in other cancer genes
Implementation of the Trusight cancer panel did not reveal any additional germline LoF mutations in 93 cancer genes among RAD51D mutation carriers.
Classification of variants of unknown significance
During the present study, we identified six VUS, each detected once. Of these, five were missense variants (p.Asn138His, p.Arg145His, p.Arg266Cys, Gly289Ser, and p.Ile311Asn), whereas one was an intronic 39 base-pair duplication (c.738 + 46dup39). All five missense variants have been previously reported in the public human variation databases (ClinVar, 1000Genomes, dbSNP), whereas the intronic variant is novel. Their possible pathogenicity has been assessed by in silico tools, already described in detail. Table 2 summarizes the in silico results, as well as clinical characteristics and family history of patients carrying these variants.
Using the protein structure modeling software, these variants have been assessed. More specifically, according to the Swiss-Model server RAD51D homology model (Fig. 3), Arg266 is solvent exposed and does not appear to make any interactions that would be perturbed when mutated to a cysteine (Fig. 3d). However, RAD51 exists as a hexamer [34] and when aligning RAD51 to the RAD51D model, Arg266 is within interaction range with Glu186 in the adjacent monomer, indicating that Arg266 may participate in inter-molecular interactions that would be perturbed in the Arg266Cys mutation. Furthermore, a solvent-exposed cysteine residue can also form disulfide bonds with other free cysteine residues leading to erroneous heterodimerization.

Mapping and interaction analysis of mutated amino acids on a RAD51D homology model. a Schematic representation of the RAD51D homology model (cyan cartoon) on which the locations of mutations are indicated (in spheres, carbon atoms in yellow, nitrogen in blue, oxygen in red). b–f Atomic interactions that the wild-type amino acids participate in, which may be disrupted by the mutations
The Ile311 residue is partially buried in a hydrophobic region, making several interactions with neighboring residues (Fig. 3f). Substitution with an asparagine residue (Ile311Asn) may promote some local destabilization, as asparagine is a hydrophilic residue—incompatible with a hydrophobic pocket—although small structural reconfigurations could allow asparagine to form hydrogen bonding interactions with nearby Arg291 or Gln115. Asn138 and Arg145 make several hydrogen bonding or electrostatic interactions with neighboring amino acids contributing to the structural stability of the molecule (Fig. 3b, c). However, mutating Asn138 and Arg145 to His may affect these interactions but to a limited degree. According to Swiss-Model server, Gly289 is located at the base of an extended loop (Fig. 3e); although it makes no specific interactions, its inherent structural flexibility is consistent with its location at the loop. The Gly289Ser variant may impose some destabilization in that loop, although it is not likely that there will be a measurable negative effect on the protein structure.
Discussion
Although the post-BRCA era unfolded through multiple efforts on the quest to discover the so-called “missing heritability,” many genes were characterized as predisposing to BC and/or OC with most of them sharing a role in the homologous recombination DNA repair pathway alongside BRCA1 and BRCA2 [35]. As mutations in these new genes are individually rare and penetrance is significantly lower than the estimated for the BRCA genes, numerous studies are needed to clarify their exact role in cancer predisposition and to justify tailored clinical interventions.
In the present study, we assessed the frequency of RAD51D mutations in a large cohort of unselected Greek OC patients, as well as in a large cohort of Greek BC patients, diagnosed before the age of 55 years and having BC or OC family history. We identified protein truncating pathogenic alterations in one OC and three BC patients, accounting for a mutation prevalence of 0.16% and 0.53%, respectively.
The mutation found in the OC patient is a novel splice acceptor site alteration (c.738 + 1 G > Α), which has been shown here to affect splicing. It is noteworthy that this patient’s disease presents the typical high-grade serous histology associated with BRCA1 and BRCA2 mutations and a defective homologous recombination mechanism [36]. All three mutations found in the BC patients have been reported before in the literature. A BC patient from a heavily burdened family with six additional BC cases (five before the age of 50 years) and one OC case carries p.Gln151Ter, which was detected before in another OC patient [12]. RAD51D p.Arg186Ter has been reported multiple times in the context of BC-OC families and may represent a British founder [10, 11, 33, 37]. Here, it was diagnosed in a 44-year-old triple-negative BC patient with two more relatives with BC under 50 years. Finally, RAD51D p.Arg300Ter, found here in a 33-year-old BC patient with two paternal aunts with BC, was previously detected in a triple-negative BC patient [13] and a patient with high-grade serous OC [10, 11, 33, 37]. A founder effect for any of the above variants, especially for the novel c.738 + 1 G > A, cannot be excluded; however, additional carrier families have to be identified in order for a haplotype analysis to be possible.
Loveday et al. [9] report a 0.88% frequency of RAD51D mutations in OC patients; however, their cohort was selected for family history [9]. In the same report they estimate a ~ 0.6% frequency in unselected OC cases, which is in line with several later studies assessing unselected OC cases and reporting mutation frequencies from 0.83% to 0.31% [2, 12, 38, 39]. The markedly lower frequency of 0.16% (one carrier among 609 unselected OC patients) found in the present study is not easily explained if no selection bias was asserted in the previous cohorts. However, we must note that the rarity of RAD51D carriers translates to weak statistical power making frequency estimations less accurate. Alternatively, the deviation noted here could represent a population-specific phenomenon. It should also be mentioned that our current screening method does not allow detection of large genomic deletions or duplications affecting complete exons or the whole RAD51D gene.
All above mentioned studies found no RAD51D mutations in BC cases, even in the context of affected families. This is surprising, considering several other studies that followed: one deleterious founder mutation (c.576 + 1 G > A) was detected in 2.9% of Finnish patients with BC and OC cancer family history [40]. A Spanish study found a 0.82% mutation frequency in familial cases of BC patients [41]. More importantly, more recent large studies reporting data from panel testing on ~ 100,000 BC patients have associated RAD51D mutations with BC, whereas reporting higher prevalence in triple-negative BC cases [13,14,15, 42]. Specifically, Buys et al. [14] screened more than 35,000 BC patients fulfilling National Comprehensive Cancer Network (NCCN) referral criteria and found 19 RAD51D mutations, which account for an overall frequency of 0.05%, but of 0.13% for triple-negative BC cases. Couch et al. [13, 15] report a similar overall frequency of 0.07% in their large study of about 65,000 BC patients. Therefore, it is of no surprise that we detected here a much higher frequency of RAD51D mutations (0.53%, 3/569 BC patients), considering the selection criteria we implemented, aiming—as already mentioned—to define a higher-risk group with a greater probability of hereditary etiology. Of particular interest is that the total of four carrier families presented here had 13 BC cases (seven before the age of 50) and only 3 OC cases. Our observation would have been enhanced if both segregation analysis and tumor analysis for loss of heterozygosity were feasible. Unfortunately, informative family members were deceased or unwilling to be tested, whereas tumor material for mutation carriers was not available.
The elevated frequency of RAD51D mutations among BC cases displayed in our study has selection bias; our ascertainment criteria for the BC cohort included family history for both BC and OC, thus favoring BC association. It must be noted, although, that only one OC case was present, compared with 13 BC cases, among families of the 3 BC patients carrying mutations (Fig. 1).
The importance of family history is depicted by the fact that no pathogenic variants were detected in patients reporting absence of family history. Our total BC cohort falls out of this category according to one of the selection criteria, however 80% of the OC cohort (485 patients) are apparently sporadic cases. With this point in mind, the frequency of RAD51D mutations in OC patients with family history (defined as having one additional relative with BC below 50 years or OC) is 0.8% compared with the 0.16% for unselected OC cases and comparable to that reported initially by Loveday et al. [9] for a similar cohort.
Penetrance estimation for moderate risk genes is a challenging task. Based on published risk estimates of 12% for an OC diagnosis by age 70 (RR (relative risk) = 6.3) [2, 38], current NCCN guidelines direct to consideration of risk-reducing salpingo-oophorectomy in premenopausal women carrying a RAD51D mutation [43]. No guidelines exist at the moment for the management of BC risk. However, in the light of the recent data by Couch et al. [13, 15] attributing a considerable moderate BC risk to RAD51D mutation carriers (OR, 3.07; 95% CI, 1.21–7.88), larger than that calculated for the ATM or CHEK2 genes for which such guidelines exist, this is a point to be considered. Further studies and meta-analyses of published data from various populations worldwide are needed to accurately estimate the penetrance of RAD51D mutations so that mutation carriers will be managed convincingly with respect to their BC and OC risk.
RAD51D inactivation has been shown to confer susceptibility to olaparib similar to that of BRCA2 inactivation [9]. Poly ADP-ribose polymerase (PARP) inhibitors impede single-stranded DNA repair, forcing cells to use homologous recombination pathway in order to mend these breaks. Hence, cells with a defective HR pathway, such as cells who have lost the function of RAD51D protein due to the presence of a germline pathogenic mutation and somatic LOH (loss-of-heterozygosity), will be unable to repair their genome and undergo apoptosis via a “synthetically lethal” response [44]. Thus, RAD51D mutation carriers may benefit from the administration of PARP inhibitors, as do patients with inactivating mutations in other HR genes such as BRCA1 or BRCA2. This treatment might improve progression-free survival among these patients.
In conclusion, our data are consistent with previous studies suggesting that RAD51D testing should be offered to affected women with a familial history of BC/OC irrespective of the number of OC cases in the family [41], and that comprehensive assessment of BC and OC risk should include evaluation of RAD51D in a multigene panel. The possible association of RAD51D mutations to BC is depicted here, adding evidence, which altogether might prompt important changes in the current surveillance guidelines. Despite their apparent rarity, determining RAD51D mutation status is important for the female relatives of affected patients, as this knowledge may allow them to make informed decisions about preventive options. Analysis of even more patient series representing various populations will be needed in order to better assess the role of the RAD51D gene in BC and OC susceptibility.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Wickramanayake A, Bernier G, Pennil C, Casadei S, Agnew KJ, Stray SM, et al. Loss of function germline mutations in RAD51D in women with ovarian carcinoma. Gynecol Oncol. 2012;127:552–5.
Ramus SJ, Harrington PA, Pye C, DiCioccio RA, Cox MJ, Garlinghouse-Jones K, et al. Contribution of BRCA1 and BRCA2 mutations to inherited ovarian cancer. Hum Mutat. 2007;28:1207–15.
Antoniou AC, Gayther SA, Stratton JF, Ponder BA, Easton DF. Risk models for familial ovarian and breast cancer. Genet Epidemiol. 2000;18:173–90.
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:18032–7.
Castera L, Krieger S, Rousselin A, Legros A, Baumann JJ, Bruet O, et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet. 2014;22:1305–13.
Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, et al. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001;15:3296–307.
Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21:2858–66.
Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43:879–82.
Osher DJ, De Leeneer K, Michils G, Hamel N, Tomiak E, Poppe B, et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br J Cancer. 2012;106:1460–3.
Thompson ER, Rowley SM, Sawyer S, kConfab, Eccles DM, Trainer AH, et al. Analysis of RAD51D in ovarian cancer patients and families with a history of ovarian or breast cancer. PLoS ONE. 2013;8:e54772.
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2:482–90.
Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33:304–11.
Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017;123:1721–30.
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–6.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9.
Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305.
Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–21.
ExAC Browser (Beta). Exome Aggregation Consortium. http://exac.broadinstitute.org. Accessed 23 February 2018.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67.
Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–508.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–94.
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–52.
Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–91.
Tomancak P, Berman BP, Beaton A, Weiszmann R, Kwan E, Hartenstein V, et al. Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2007;8:R145.
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–8.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105–17.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing Exploratory research consortium. Am J Hum Genet. 2016;98:1067–76.
Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol. 2015;33:2901–7.
Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, et al. Crystal structure of a Rad51 filament. Nat Struct Mol Biol. 2004;11:791–6.
Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5.
Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10:803–8.
Baker JL, Schwab RB, Wallace AM, Madlensky L. Breast cancer in a RAD51D mutation carrier: case report and review of the literature. Clin Breast Cancer. 2015;15:e71–5.
Lilyquist J, LaDuca H, Polley E, Davis BT, Shimelis H, Hu C, et al. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol. 2017;147:375–80.
Harter P, Hauke J, Heitz F, Reuss A, Kommoss S, Marmé F, et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1). PLoS ONE. 2017;12:e0186043.
Pelttari LM, Kiiski J, Nurminen R, Kallioniemi A, Schleutker J, Gylfe A, et al. A Finnish founder mutation in RAD51D: analysis in breast, ovarian, prostate, and colorectal cancer. J Med Genet. 2012;49:429–32.
Gutierrez-Enriquez S, Bonache S, de Garibay GR, Osorio A, Santamariña M, Ramón y Cajal T, et al. About 1% of the breast and ovarian Spanish families testing negative for BRCA1 and BRCA2 are carriers of RAD51D pathogenic variants. Int J Cancer. 2014;134:2088–97.
Ollier M, Radosevic-Robin N, Kwiatkowski F, Ponelle F, Viala S, Privat M, et al. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. Am J Cancer Res. 2015;5:2113–26.
Daly MB, Pilarski R, Berry M, Buys SS, Farmer M, Friedman S, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast and ovarian, version 2.2017. J Natl Compr Canc Netw. 2017;15:9–20.
Banerjee S, Kaye SB, Ashworth A. Making the best of PARP inhibitors in ovarian cancer. Nat Rev Clin Oncol. 2010;7:508–19.
Acknowledgements
We thank the patients and their families for their participation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Konstanta, I., Fostira, F., Apostolou, P. et al. Contribution of RAD51D germline mutations in breast and ovarian cancer in Greece. J Hum Genet 63, 1149–1158 (2018). https://doi.org/10.1038/s10038-018-0498-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0498-8
