
Abstract
Genome-wide association studies (GWAS) have identified many susceptibility loci for cardiometabolic disorders. Most of the associated variants reside in non-coding regions of the genome including long non-coding RNAs (lncRNAs), which are thought to play critical roles in diverse biological processes. Here, we leveraged data from the available GWAS meta-analyses on lipid and obesity-related traits, blood pressure, type 2 diabetes, and coronary artery disease and identified 179 associated single-nucleotide polymorphisms (SNPs) in 102 lncRNAs (p-value < 2.3 × 10−7). Of these, 55 SNPs, either the lead SNP or in strong linkage disequilibrium with the lead SNP in the related loci, were selected for further investigations. Our in silico predictions and functional annotations of the SNPs as well as expression and DNA methylation analysis of their lncRNAs demonstrated several lncRNAs that fulfilled predefined criteria for being potential functional targets. In particular, we found evidence suggesting that LOC157273 (at 8p23.1) is involved in regulating serum lipid-cholesterol. Our results showed that rs4841132 in the second exon and cg17371580 in the promoter region of LOC157273 are associated with lipids; the lncRNA is expressed in liver and associates with the expression of its nearby coding gene, PPP1R3B. Collectively, we highlight a number of loci associated with cardiometabolic disorders for which the association may act through lncRNAs.
Similar content being viewed by others

Introduction
Recent data from high-throughput sequencing platforms have established that most of the human genomic DNA has biochemical activity [1, 2]. These data show that the protein-coding genes make only a very small proportion (~2%) of the genome, indicating a lack of understanding in the possible contribution of non-coding elements to disease biology. Although the non-coding regions of the genome do not encode proteins, they are often transcribed into non-coding RNAs (ncRNAs) [3]. The ncRNAs can be roughly categorized, based on their transcript size, into small ncRNAs (such as microRNAs (miRNAs)) and long ncRNAs (lncRNAs) [4, 5]. The lncRNAs are defined as non-protein-coding transcripts over 200 nucleotides in length [6, 7]. Similar to messenger RNAs (mRNAs), lncRNAs are transcribed by RNA polymerase II, 5′-capped and polyadenylated [8, 9]. Moreover, many of them contain multiple exons and are subjected to alternative splicing [3, 10]. Over the past few years, it has become increasingly evident that lncRNAs act as important players in the epigenetic, post-transcriptional, and translational coordination of gene expression in various biological processes and human diseases [11,12,13]. The expression and function of a number of lncRNAs have been shown to be dysregulated in cardiovascular disease, such as MHRT in heart failure [14]. In addition, genome-wide association studies (GWAS) have reported a relationship between lncRNA genetic variants and cardiovascular disease, including variants in CDKN2B-AS (ANRIL) that are associated with the risk of coronary artery disease (CAD) and variants in MIAT that confer susceptibility to myocardial infarction [15,16,17]. Nevertheless, despite some well-characterized lncRNAs (e.g., ANRIL), little is known about the general features of most lncRNAs and their molecular mechanisms in cardiometabolic disorders.
Previous large-scale GWAS meta-analyses have enabled the discovery of a number of common genetic variants at multiple loci associated with cardiovascular disease and its risk factors [6, 18, 19]. However, the majority of the associated variants map to non-coding regions, and their biological relevance to the disease remains poorly understood [20]. The functional impact of the non-coding variants is particularly difficult to study since their functional consequences should be investigated beyond the context of protein-coding genes [21, 22]. Many studies have focused recently on the functional annotation of GWAS hits that are localized in ncRNAs [21,22,23,24,25]. We and others have shown previously that variants in miRNAs can explain some of the GWAS associations with cardiometabolic disorders [26,27,28,29]. However, to date, the association of lncRNA variants with cardiometabolic disorders has not been investigated systematically. In the present study, we aimed to identify the associations with cardiometabolic disorders in non-coding regions that may act through lncRNAs. To this end, we retrieved genetic variants located in lncRNAs and examined their associations with cardiovascular disease and its risk factors using publicly available GWAS data. We subsequently performed in silico analyses for the associated variants (e.g., expression quantitative trait loci (eQTL) analysis and annotation to regulatory features) as well as expression and DNA methylation analysis for their lncRNAs to determine lncRNAs that might play a role in cardiometabolic disorders.
Materials and methods
GWAS on cardiometabolic disorders
Summary association statistics from the recent GWAS meta-analyses on 10 cardiometabolic disorders were used for association analysis. The cardiometabolic disorders in our analysis included four lipid traits (plasma concentrations of total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides) from the Global Lipids Genetics Consortium (GLGC) [18], two anthropometric measures (body mass index (BMI) and waist to hip ratio (WHR)) from the Genetic Investigation of ANthropometric Traits (GIANT) consortium [30, 31], systolic and diastolic blood pressure from the Global BPgen consortium [32], CAD from the CARDIoGRAMplusC4D consortium [6], and type 2 diabetes (T2D) from the DIAGRAM consortium [19]. All participating studies in these GWASs had provided the informed consent for participation in genetics studies and were approved by their local ethical committees. Descriptions of the GWAS meta-analyses, the consortia responsible for these analyses, the sample sizes, and the number of variants associated with each trait are provided in Table S1.
Association of genetic variants in lncRNAs with cardiometabolic disorders
We retained variants that are located in human lncRNA transcripts using the lncRNASNP database (http://bioinfo.life.hust.edu.cn/lncRNASNP/), a comprehensive database that includes 495,729 single-nucleotide polymorphisms (SNPs) in 32,108 lncRNA transcripts of 17,436 lncRNA genes [33]. This database is based on two online resources: dbSNP (build 138) (http://www.ncbi.nlm.nih.gov/SNP/) and LNCipedia (http://www.lncipedia.org/). We analyzed 21,915 SNPs (in 8829 lncRNAs) with minor allele frequencies (MAF) >0.01 that were available in the GWASs data for association with cardiometabolic disorders. The number of statistical tests was calculated by multiplying the number of examined SNPs by the number of traits (21,915 × 10). The Bonferroni correction method was used to adjust the p-value for multiple testing and establish the study significance threshold (p-value < 2.3 × 10–7). Manhattan and regional plots showing the association of lncRNA variants and flanking variants in the corresponding loci with cardiometabolic disorders were generated by R and the LocusZoom web tool [34].
Association of lncRNA variants with expression of nearby and distant genes
The association of lncRNA variants with expression of the hosting lncRNA, and nearby and distant coding genes (eQTL) were examined in blood and adipose tissue. The eQTL from adipose tissue was used, since a high percentage of the identified variants were found to be associated with lipid traits, BMI, and WHR. To this end, data from samples of 856 healthy female twins included in the MuTHER Study were used [35]. In addition, the eQTL from whole blood of 5311 individuals, accessed through the Gene Network database (http://genenetwork.nl/bloodeqtlbrowser/), was used. This particular eQTL data set was used because of its large sample size and power [36]. For eQTL analysis of cis-eQTLs (<1 MB), the significance threshold was set by dividing the nominal p-value of 0.05 by the number of studied SNPs. For eQTL analysis of trans-eQTLs (≥1 MB), given the large number of tests (the number of lncRNA SNPs multiplied by 23,644 probes for all transcripts) and the relatively small effect sizes of trans-eQTLs, a false discovery rate (FDR) of 1% was used [35].
Annotating the regulatory features to lncRNA variants
The lncRNA variants were annotated to regulatory features, including promoter and enhancer regulatory motifs, DNase footprinting sites and conserved sequences using HaploReg v 4.0 database (http://www.broadinstitute.org/mammals/haploreg/haploreg.php). Further, for each lncRNA variant, the linkage disequilibrium (LD) region (R2 > 0.8) was determined using the 1000G Phase 1 population and proxy variants in strong LD with a given lncRNA variant were extracted. Then, we investigated whether the lncRNA variant and its proxies are located in potential regulatory regions across a number of available cell lines and tissues from Roadmap epigenomes dataset (see supplementary methods for details) [37, 38]. The reference epigenomes used were chromatin state models based on imputed data of 12 histone marks [38], identifying separate epigenetic chromatin states for the lncRNA variants. Heat map was constructed by calculating the percentage of variants in LD with the given lncRNA variant located in DNA regulatory regions as defined by the chromatin states. In addition, we examined the genes surrounding the lncRNA variants using the publicly available database, Online Mendelian Inheritance in Man (OMIM) [39], to see if any of the surrounding genes are known to be involved in Mendelian cardiovascular disorders.
Potential impact of variants on the interaction between lncRNAs and miRNAs
The lncRNASNP database was used to predict whether the lncRNA variants are located in predicted miRNA-binding sites and might potentially affect lncRNA–miRNA interactions [33]. A functional prerequisite for miRNA-dependent transcriptional regulation is that the miRNA and its targets are expressed in the same target tissue. Thus, we used multiple web tools to investigate whether lncRNAs and miRNAs in our list are expressed in relevant cardiovascular tissues (see supplementary methods for details). Additionally, we performed Luciferase reporter assays for one candidate lncRNA variant to examine the binding of lncRNA to the related miRNA, and also to determine the effect of the variant on the lncRNA–miRNA interaction. To this end, primers were designed to amplify the lncRNA sequence (wild type and mutated), the forward and reverse primers included the restriction enzyme sites XbaI and ApaI, respectively. All primers are shown in Table S2. The lncRNA sequences containing the binding site of miRNA were amplified and cloned into the pGL3 Luciferase reporter vector (Promega) downstream of the Luciferase open reading frame [40]. The inserts of all constructs were confirmed by Sanger sequencing. The HepG2 cells (n = 10,000) were plated into 96-well plates and co-transfected with 1 μg of pGL3 containing the lncRNA sequence (with either the major or minor allele), miRNA mimic (mirVana TM Mimics), and a plasmid expressing the Renilla Luciferase (as transfection control), using LipofectamineR RNAiMAX (Invitrogen, Carlsbad, CA, USA). Luciferase activity was determined using the Dual-Glo Luciferase Assay System according to manufacturer’s protocol (Promega, Madison, WI, USA). Renilla activity was used for normalization of the data. All experiments were performed in triplicate.
Prediction of the variant effect on the lncRNA secondary structure
For lncRNA variants associated with cardiometabolic disorders, we used both lncRNASNP database and RNAsnp web tool to predict the variant impact on the lncRNA secondary structure [33, 41]. In brief, the lncRNA transcript sequences were extracted from human reference genome (GRCh37/hg19) according to the lncRNA transcript BED file as reference transcripts. Then, the corresponding allele in the given reference-transcript was changed to the alternative allele as alternative-transcript. The RNAfold program (http://rna.tbi.univie.ac.at/) was subsequently used to calculate the minimal free energy (MFE, ΔG) and illustrate the lncRNA secondary structure. Energy change of RNA structures (ΔΔG) was quantified as the difference between the level of MFE of the thermodynamic ensemble of alternative vs. reference transcript (ΔΔG = ΔG alternative transcript−ΔG reference transcript).
Expression of lncRNAs in tissues relevant to cardiometabolic disorders
The Human Body Map catalog, which is one of the most complete set of expression data for lncRNAs including the RNA-seq across 22 human tissues and cell lines, was used to examine the expression of identified lncRNAs in cardiovascular tissues [42]. Moreover, we used gene expression data from 83 subcutaneous adipose tissue (SAT), 77 visceral adipose tissue (VAT), 74 liver samples, and 62 muscle skeletal samples from our cohort of 85 unrelated obese Dutch individuals [43, 44]. This analysis enabled us to detect the expression levels of lncRNAs with probe available in Illumina Human HT12v3 microarray. Real-time PCR was used to measure the expression of a candidate lncRNA in human liver cell line (H7).
Association of DNA methylation and expression of lncRNAs with cardiometabolic disorders
We extracted the cytosine–phosphate–guanine (CpG) sites within (+/−1 kb) the identified lncRNAs and examined the association of DNA methylation levels at these CpG sites with cardiometabolic disorders using data from the Rotterdam Study (RS). The design of the RS has previously been described elsewhere [45]. Briefly, the RS is a prospective, population-based cohort study that comprises three cohorts. The initial cohort (RS-I) started out in 1990 with 7983 participants aged 55 years and over from the neighborhood Ommoord in Rotterdam. In 2000–2001, a second cohort was established (RS-II) with 3011 inhabitants. The third cohort (RS-III) started in 2006 with 3932 inhabitants aged 45 years and over. In the current study, we used the methylation data from a set of 767 participants from the third visit of the second cohort (RSII-3) and the second visit of the third cohort (RSIII-2) as the discovery panel (see Supplementary Methods for details). Moreover, we sought replication in a set of 731 participants from RSIII-1 with DNA methylation data available [36, 46]. None of the participants included in the replication study were included in the discovery cohort. Methylation levels were measured in whole blood using the Illumina Methylation 450 array. The associations between cardiometabolic disorders and DNA methylation β-values were examined using linear mixed-effect models. All models were adjusted for sex, age, smoking, white blood cell proportions, array number, and position on the array. The baseline characteristics of the participants in the study are shown in Table S3. In addition, we examined the association between the expression levels of the identified lncRNAs (with probe available in the Illumina Human HT12v3 microarray) and cardiometabolic disorders using gene expression data from a random subset of 881 individuals in RSIII-1 [36]. A linear regression model was used to examine the association between the traits and lncRNA expression levels, adjusting for age, sex, BMI, RNA Quality Score, plate ID (batch), and cell counts.
Results
lncRNA variants associated with cardiometabolic disorders
A flowchart of our approach to identify lncRNAs associated with cardiometabolic disorders is visualized in Fig. 1. We examined the associations of 21,915 SNPs (with MAF > 0.01 and available in the GWASs data) in 8829 lncRNAs with lipid traits, BMI, WHR, systolic and diastolic blood pressure, T2D, and CAD. Manhattan plots showing the associations of these lncRNA SNPs and cardiometabolic disorders are presented in Figure S1. We found significant associations between 179 unique SNPs in 102 lncRNAs and the studied traits (p-value < 2.3 × 10–7) (Table S4). Out of the 179 associated SNPs, 90 SNPs are located in long intergenic ncRNAs (lincRNAs), 53 in intronic lncRNAs, and 36 in other lncRNA subtypes, including antisense transcripts. Ninety-nine of 102 lncRNAs hosting the 179 associated SNPs are located in the loci that have been reported in the original GWASs to be associated with cardiometabolic disorders [6, 18, 19, 30,31,32]. In addition, we found three loci suggestively associated with cardiometabolic disorders (p-value between 5 × 10–8 and 2.3 × 10–7), that need to be replicated in future association studies. The lncRNA SNPs in these new loci include rs709400 (lnc-XRCC3-1, chr14: 103683138) associated with BMI, rs7537211 (lnc-CACNA1E-2, chr1: 182089421) associated with HDL, and rs9355860 (lnc-MAP3K4-1, chr6:160969755) associated with LDL (Table S5). Among the 179 associated SNPs, 17 were lead SNP showing the strongest association with the traits (Figure S2) and 38 were in strong LD (R2 > 0.8) with the lead SNP in the related loci. We focused on these 55 variants for our further investigations (Table S6). We calculated variance explained by the 55 lncRNA variants in the RS that are shown in Table S6.

Identification of lncRNAs associated with cardiometabolic disorders. This flowchart shows our approach to identify lncRNA SNPs that are associated with cardiometabolic disorders and to provide additional evidence supporting the identfied associations. GWAS genome-wide association studies, SNP single-nucleotide polymorphism, eQTL expression quantitative trait loci, MAF minor allele frequency, LD linkage disequilibrium, miRNA microRNA, lncRNA long non-coding RNA, CMD cardiometabolic disorders. (Color figure online)
lncRNA variants associated with gene expression
The cis-eQTL data were used to determine the correlation between the 55 selected SNPs and expression levels of their nearby genes (<1 MB). The significance threshold for this analysis was set at p-value < 9.1 × 10–4 (0.05/55). Twenty-seven SNPs were found to be significantly associated with the expression levels of their nearby genes either in blood (26 SNPs) (Table S7) or in adipose tissue (17 SNPs) (Table S8). Out of the 17 SNPs with cis-eQTL in adipose tissue, 16 SNPs showed the eQTL also in blood, while rs709400 in lnc-XRCC3-1 associated with BMI showed eQTL only in adipose tissue. Some of the eQTL SNPs were associated with the expression levels of multiple nearby genes (sometimes in both tissues) including rs3099844 in linc-HLA-B-1, rs3177647 in lnc-TMEM116-2 (also known as linc-BRAP-2), rs9925964 in lnc-BCKDK-1, rs9934328 in lnc-ARGP-2, rs2301826 in lnc-RCCD1-1, rs2898290 in lnc-BLK-1, and rs6760828 in lnc-SNX17-1.
We also checked the correlation between the 55 selected SNPs and 23,644 probes for all transcripts (trans-eQTL) in adipose tissue and whole blood. We used an FDR of 1%, which corresponded to p-value < 2.0 × 10–6 in adipose tissue and p-value < 1.5 × 10–7 in blood. Four SNPs were found to be significantly associated with the expression levels of distant genes in the adipose tissue, including rs17427875 (lnc-EVX1-5) with C19orf12, rs2070959 (lnc-HJURP-2) with PRDM1, rs456598 (lnc-SLC22A2-1) with BPIL1 and UCP3, and rs7537211 (lnc-CACNA1E-2) with PHF21A and ZNF746 (Table S9). Two SNPs were also significantly associated with the expression levels of distant genes in blood, including rs243018 (lnc-BCL11A-3) with ESPN and rs3099844 (lnc-ATP6V1G2) with TMEM154, BTN3A2, and HIST1H2BD (Table S9).
In silico predictions and regulatory annotations for the 55 selected lncRNA variants
Using HaploReg v4.0, we showed that most of the 55 selected lncRNA SNPs were not in strong LD with any non-synonymous variants in the corresponding loci (Table S10). Next, for each set of proxy variants in high LD (R2 > 0.8) with the lncRNA SNPs, we examined if the variants are located in a potential regulatory region and are enriched in specific cell type relevant to cardiometabolic disorders (Table S10). This analysis indicated that almost all SNPs are not over-represented in the regulatory regions related to the associated cardiometabolic disorders. Rs4841132 in lnc-RP11-10A14.4 (LOC157273), associated with lipid traits, was the only SNP that the (potential) regulatory region, in which the SNP is located, was enriched in liver, which is the relevant cell type (Fig. 2).

Heat map of the enrichment of lncRNA SNPs in the potential regulatory regions. This heat map was constructed by calculating the percentage of 55 lncRNA SNPs and their proxies in strong LD (R2 > 0.8) located in DNA regulatory regions as defined by the chromatin states using the Roadmap epigenomes data set across tissues relevant to cardiovascular disease (e.g., aorta, fetal heart, atrium, and ventricle), metabolic disorders (e.g., liver, pancreas, and adipose), and also control tissues (e.g., lung and brain). The lncRNA SNPs which are top variants in their loci associated with cardiometabolic disorders are shown in bold. As shown, almost all SNPs are not over-represented in the regulatory regions related to cardiometabolic disorders. Rs4841132 in lnc-RP11-10A14.4 (LOC157273) was the only SNP that the (potential) regulatory region, in which the SNP is located, was enriched for the relevant cell type (liver). (Color figure online)
It has been shown that lncRNAs could indirectly regulate the expression levels of coding genes either by acting as miRNA decoys [33, 47] or as competitors for miRNAs by binding to their target genes [48]. Our analysis showed that 25 out of the 55 selected SNPs are localized in sequences that match to the seed sequence of miRNAs (Table S11). However, lncRNAs can only function as molecular sponges for miRNAs, if the localization of the lncRNA is appropriate and the lncRNA expression level is sufficiently high [49, 50]. We thus tested the expression levels of lncRNAs (harboring SNPs in predicted miRNA-binding sites), with probe available in the Illumina microarray, in cardiovascular tissues using gene expression data from our cohort of 85 Dutch individuals [43, 44]. We found five lncRNAs that are abundantly expressed in adipose tissue, liver, and skeletal muscle (Table S12). The RNA-seq data from the Human Body Map catalog then showed another four lncRNAs expressed in cardiovascular relevant tissues (Figure S3). To identify evidence for the expression of the interacting miRNAs in cardiovascular tissues, we used multiple miRNA expression databases. The lncRNAs and miRNAs expressed in cardiovascular tissues that their interactions might be affected by the identified lncRNA SNPs are highlighted in Table S11.
The main functions of lncRNAs include interaction with DNA, RNA, and protein. The secondary structure is thereby important for the lncRNA functions. Previous studies have shown correlations between the presence of variants in lncRNAs that affect the secondary structure and expression of the lncRNAs, and the development of specific diseases [51,52,53]. We thus checked whether the 55 selected SNPs might affect the secondary structure of lncRNAs. Differences in MFE (ΔMFE > 0.1) of the thermodynamic ensemble of mutant vs. wild-type lncRNA sequences were observed for 48 out of the 55 SNPs (Table S13). It can be hypothesized that the disruption of lncRNA secondary structure by these SNPs might change the processing or function of the hosting lncRNAs [51].
Association of DNA methylation and expression of the lncRNAs with cardiometabolic disorders
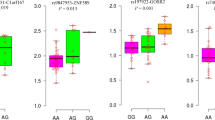
We found 518 CpG methylation sites annotated to 38 of the 55 lncRNAs, hosting the associated SNPs, and examined their association with cardiometabolic disorders using the RS data. Table 1 displays the top 10 CpGs showing the most significant association with the studied traits, which are in line with the observed associations in the GWAS data. After applying the Bonferroni correction, the significance threshold of 9.65 × 10–5 (0.05/518), only one CpG remained significant. This includes cg17371580 annotated to the promoter region of LOC157273 associated with HDL cholesterol (meta-analysis p-value = 3.65 × 10–5, effect = 0.0094). The CpG sites with a nominal association with cardiometabolic disorders are shown in Table S14.
Next, we tested the association between expression level of 13 of the 55 identified lncRNAs, with probe available in the Illumina Human HT12v4 microarray, and cardiometabolic disorders using the RS data. After applying the Bonferroni correction, the significance threshold of 3.85 × 10–3 (0.05/13), we found no significant association (Table S15). The expression levels of two lncRNAs were nominally associated with the studied traits, including lnc-IER3-5 (ILMN_1657996) associated with total cholesterol (p-value = 4.65 × 10–3, effect = −4.09) and lnc-MTAP-1 (also known as CDKN2B-AS) (ILMN_2376723) associated with CAD (p-value = 1.24 × 10–2, effect = 0.42).
Prioritizing the identified lncRNA variants associated with cardiometabolic disorders
We combined the results of our analyses for the 55 selected lncRNA SNPs and prioritized them based on their potential functionality in relation to cardiometabolic disorders; a summary overview of our findings for the prioritized SNPs is shown in Table 2 and Table S16. The SNP with the highest credibility for functionality in this list is rs4841132 located in LOC157273 (Fig. 3). As one of the prediction for this SNP was to affect the binding of LOC157273 to miR-1287, we performed Luciferase reporter assay to first test the regulatory interaction between these two ncRNAs, and then examine the SNP effect on the lncRNA–miRNA interaction. Co-transfection of LOC157273 luciferase constructs with miR-1287 mimic in Hep2G cells showed no significant difference between the luciferase activities of the constructs containing the major and minor alleles of rs4841132 (Figure S4). The negative result can be explained by compensation of the miRNA–lncRNA binding outside the miRNA seed sequence [54]. In addition, LOC157273 has three binding sites for miR-1287, which rs4841132 is predicted to affect one of them. These data may indicate that rs4841132 is unlikely to act through affecting the lncRNA–miRNA interaction.

Genetic and functional evidence suggesting LOC157273 to be a lipid susceptibility gene. a The regional association plots show the association of variants in LOC157273 with HDL and LDL cholesterol. b A figure generated by the UCSC Genome Browser on 8p23.1 locus hosting LOC157273. The position of SNP rs4841132 within the second exon of LOC157273 and CpG site cg17371580 within the conserved promoter region of lncRNA are depicted by red arrows. The figure also illustrates the expression of LOC157273 across different tissues using the Human Body Map catalog, indicating that the lncRNA is expressed in liver. c A schematic showing the lncRNA structure, the nearest protein coding gene PPP1R3B, the position of SNPs and a CpG site in the lncRNA associated with lipid traits. (Color figure online)
Discussion
In a systematic investigation of lncRNAs associated with cardiometabolic disorders, we identified 179 associated SNPs in 102 lncRNAs. We selected 55 of the SNPs (either the lead SNP or in strong LD with the lead SNP in the related loci) and performed various in silico analyses for the SNPs as well as expression and DNA methylation analysis for their hosting lncRNAs in relation to cardiometabolic disorders. Our results showed a number of lncRNA SNPs (e.g., rs4841132 in LOC157273) that fulfilled predefined criteria for being potentially functional variants in their loci, emphasizing the importance of the associated lncRNAs in the genetic susceptibility of cardiometabolic disorders.
We found 179 lncRNA SNPs significantly associated with cardiometabolic disorders. Many of these SNPs were localized adjacent to protein-coding genes. Due to the close proximity to the coding regions, it was difficult to ascertain whether the observed GWAS association is driven by the lncRNA SNP or its proxy coding variants. We therefore limited the set of lncRNA SNPs to those that are either the sentinel SNP or one of the top SNPs in high LD with the sentinel SNP in the corresponding loci associated with cardiometabolic disorders. This filtration was applied since in case of a single functional variant, the functional SNP is expected to show the strongest association with the trait. In case of multiple functional variants in one locus, we should have either identified multiple independent signals or find the functional variants within the top SNPs [55]. Hence, the filtration enriched the set of associations for those that are more likely to be mediated by lncRNAs. Notably, the majority of the 55 selected SNPs that remained after this filtration had no non-synonymous proxy variants in high LD in their vicinity.
In order to identify whether the 55 selected SNPs could functionally affect the lncRNAs, several criteria were adopted. An established association between the variant and a trait, the correlation of variant with expression of the lncRNA or protein-coding genes, the localization of variant in regulatory regions, and the potential of variant for structural perturbations within the lncRNA are a number of such criteria [25, 33, 49, 56]. These criteria mainly assess the functionality of SNPs and extend them to the host lncRNA. As a second step, we investigated whether the lncRNAs hosting the 55 selected SNPs are related to the trait of interest by performing expression and DNA methylation analyses. Together, these analyses enabled us to highlight a number of genetic associations that might affect cardiometabolic disorders through lncRNAs. At least some of these associations have been incorrectly annotated in the literature, where the nearest protein-coding gene to the SNP is reported as the trait-associated gene, while the lncRNA directly encompassing the SNP is ignored or the mediatory role of lncRNA is overlooked. We discuss some of these lncRNA SNPs associated with cardiometabolic disorders below.
The first SNP is rs4841132 in chromosome 8 locus (8p23.1) that has been reported by a number of GWAS to be associated with lipid levels, fasting insulin, fasting glucose, and obesity [18, 57,58,59,60]. The association was annotated to PPP1R3B in the literature. Here, we found a CpG site within the conserved promoter region of LOC157273 that is significantly associated with serum HDL cholesterol. The lead SNPs in 8p23.1 locus are rs4841132 and rs9987289, which are in perfect LD (R2 = 1). Rs4841132 is located in the second exon of LOC157273, the nearest protein-coding gene is PPP1R3B (~175 kb away) and its variants are not in LD with the lncRNA SNP. Our fine mapping further showed that rs4841132 is mapped to promoter histone marks in liver, a relevant tissue for lipid metabolism. The second SNP, rs9987289, resides in the first intron of the lncRNA and is a cis-eQTL for PPP1R3B in human liver [57, 61]. In addition, Lipovich et al. have reported more recently that LOC157273 knockdown increased the PPP1R3B mRNA levels in hepatocytes, indicating that the lncRNA represses PPP1R3B expression [62, 63]. PPP1R3B is a relevant gene for the associated traits (glycemic and lipid metabolic disorders) since it encodes protein phosphatase 1 regulatory subunit 3B, which inhibits glycogen breakdown by regulating the interaction of phosphorylated protein 1 (PP1) with glycogen metabolism enzymes [64]. LOC157273 is also shown to be expressed in human hepatocytes [42]. These data together may suggest LOC157273 to be the key regulator that links the identified variants with PPP1R3B activity and with lipid profile.
The second SNP is located in the well-known KCNQ1 locus on chromosome 11 (11p15.5) associated with T2D [65]. The top hit in this locus, rs231361, overlaps both the lnc-CDKN1C-3 (also known as KCNQ1OT1) transcript and intron 11 of KCNQ1. The KCNQ1 gene has been reported in several studies to be associated with cardiovascular events and T2D [66,67,68,69,70,71,72]. This gene encodes a voltage-gated potassium channel and plays an important role in repolarization of the cardiac action potential [73, 74]. Korostowski et al. demonstrated that the expression level of KCNQ1 gene is upregulated in mice expressing a truncated KCNQ1OT1, indicating that the lncRNA represses KCNQ1 expression [75]. Moreover, two independent studies have revealed that KCNQ1OT1 negatively regulates the expression of CDKN1C, a known regulator of beta-cell development [76, 77]. Further, the SNP rs231361 has been shown to influence the expression of nearby genes including CDKN1C [78, 79]. These data suggest that rs231361-induced aberrant expression of KCNQ1OT1 may be sufficient to cause disease through deregulation of the expression levels of KCNQ1 and CDKN1C, two coding genes that are most likely to mediate T2D susceptibility at the KCNQ1 locus.
The third SNP is located in chromosome 6 harboring IER3-5 (also known as LINC00243) associated with triglyceride and total cholesterol. In our study, we found that the expression level of LINC00243 is nominally associated with total cholesterol. In addition, we observed that methylation of a CpG site annotated to this lncRNA is nominally associated with triglyceride. The lead variant in this locus, rs886424, resides in the second exon of LINC00243, ~70 kb away from IER3 that was thought to be the disease candidate gene. Through the eQTL analysis, we observed a correlation between rs886424 and the expression level of LINC00243 and its nearby coding genes including IER3, VARS2, FLOT1, MICB, HLA-C, HLA-H, and HLA-G. Performing a pathway analysis (using IPA) with these genes, we found that they are enriched in the immunological disease pathway. In particular, IER3 has been reported to inhibit the production of the pro-inflammatory cytokines [80]. Since lncRNAs have been suggested to modulate the same biological processes as regulated by their nearby genes [81,82,83], LINC00243 can be hypothesized to play a regulatory role in immune-function-related pathways that need to be confirmed in further experimental studies.
While the above-mentioned examples endorsed the annotation in the literature, however, suggests a lncRNA to mediate the observed association, another part of our findings indicate potential incorrect annotations to nearby coding genes. For instance, rs17427875 was found to be associated with WHR and was annotated to HOXA11, which is a nearby coding gene and ~95 kb downstream of the variant. Nevertheless, this sentinel SNP resides within the first exon of HOXA11 antisense transcript (HOXA11-AS). This lncRNA is located in the 5′ region of the HOXA gene cluster, is highly conserved amongst several species [84], and is abundantly expressed in adipose tissue [42]. Previous studies have suggested that antisense transcript lncRNAs can directly or indirectly regulate the expression of the sense genes; however, experimental studies for HOXA11-AS have shown no effect on HOXA11 expression or function [85, 86]. Instead, Sun et al. revealed that knockdown of HOXA11-AS upregulates KLF2, which is a well-known anti-adipogeneic gene, through functioning as a scaffold to control the gene expression [89]. In addition, our cis-eQTL data showed a correlation between rs17427875 and expression level of HOXA5, a nearby gene with an established association with the obesity-related phenotypes [87,88,89]. Through trans-eQTL analysis in adipose tissue, we further observed a correlation between rs17427875 and the expression level of C19orf12 (also known as NBIA4), which is itself shown to be upregulated during adipocyte differentiation [90]. Together, these data suggest a role for HOXA11-AS in fat distribution that may take place through regulating the expression of the aforementioned coding genes, and not HOXA11.
Likewise, rs243018 is a lead SNP associated with T2D in an evolutionary conserved locus on chromosome 2 (2p16.1). The variant is localized in the last exon of lnc-BCL11A-3, which overlaps with miR-4432 host gene. However, the genetic association is annotated to the nearest coding gene, BCL11A, in the literature and the miRNA host gene that directly encompass the variant is overlooked [91, 92]. BCL11A gene is upstream (~90 kb away) of rs243018 and its variants are not in LD with the variant. The whole blood tran-eQTL data showed the association of rs243018 with the expression of ESPN, a gene which is linked previously to Insulin-like growth factor 1 [93].
This study has strengths as well as limitations that should be considered with the interpretation of the reported results. The availability of summary statistics from the largest GWAS meta-analyses of cardiometabolic disorders enabled us to detect multiple lncRNAs associated with the studied traits. In addition, the availability of genome-wide DNA methylation and expression data in the RS allowed us to further investigate the potential causal mechanisms in the identified regions. Our study, however, has some limitations that need to be addressed. Although we had a reasonable sample size for methylation and expression analysis, weaker associations could have been overlooked due to lack of power. Another limitation is the use of whole blood samples to determine DNA methylation and expression levels of the lncRNAs, which is not the most relevant tissue. DNA methylation is cell type specific. LncRNA transcripts, on average, express at lower levels and show a higher degree of tissue specificity in comparison to protein-coding transcripts. Further, many of the identified lncRNAs had either no probe available in the Illumina expression array or the probes did not pass quality control; we thus could not investigate the association between expression levels of these lncRNAs and cardiometabolic traits. Although this could have resulted overlooking some of the lncRNAs, the found associations are valid. In an optimal setting, one should examine the associations using expression arrays covering all lncRNAs or next-generation sequencing in target tissues (e.g., adipose tissue, heart, and liver). Such infrastructure is not yet available in large epidemiologic studies with validated clinical data.
In conclusion, the present study provides a systematic investigation of lncRNAs associated with cardiometabolic disorders. We found a number of cardiovascular-associated loci for which the lead variant lies in an lncRNA that is likely to contribute to the pathogenesis of the associated disorder. In particular, we found evidence suggesting LOC157273 (at 8p23.1) to be involved in regulating serum lipid-cholesterol. The identified lncRNAs are candidates for future studies to determine their functions at different stages of cardiac development and metabolic processes, as well as their potential as diagnostic biomarkers and therapeutic targets for cardiovascular disease and its risk factors.
References
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63.
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Pennisi E. Genomics. ENCODE project writes eulogy for junk DNA. Science. 2012;337:1159.
ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816.
CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8.
Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27.
Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253–61.
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–61.
Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–6.
Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–56.
Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17:806–14.
Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011;25:444–8.
The Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–44.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7.
Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24:R102–10.
Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11:294–6.
Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 2012;30:1095–106.
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–20.
Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–97.
Huan T, Rong J, Tanriverdi K, Meng Q, Bhattacharya A, McManus DD, et al. Dissecting the roles of microRNAs in coronary heart disease via integrative genomic analyses. Arterioscler Thromb Vasc Biol. 2015;35:1011–21.
Ghanbari M, Sedaghat S, de Looper HW, Hofman A, Erkeland SJ, Franco OH, et al. The association of common polymorphisms in miR-196a2 with waist to hip ratio and miR-1908 with serum lipid and glucose. Obesity. 2015;23:495–503.
Ghanbari M, de Vries PS, de Looper H, Peters MJ, Schurmann C, Yaghootkar H, et al. A genetic variant in the seed region of miR-4513 shows pleiotropic effects on lipid and glucose homeostasis, blood pressure, and coronary artery disease. Hum Mutat. 2014;35:1524–31.
Li Q, Chen L, Chen D, Wu X, Chen M. Influence of microRNA-related polymorphisms on clinical outcomes in coronary artery disease. Am J Transl Res. 2015;7:393–400.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Magi R, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–96.
International Consortium for Blood Pressure Genome-Wide Association S, Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9.
Gong J, Liu W, Zhang J, Miao X, Guo AY, lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. 2014;43:D181–6.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7.
Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9.
Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–43.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8.
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30.
McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604.
Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM, et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood. 2011;118:916–25.
Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. RNAsnp: efficient detection of local RNA secondary structure changes induced by SNPs. Hum Mutat. 2013;34:546–56.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9:e1003201.
Fu J, Wolfs MG, Deelen P, Westra HJ, Fehrmann RS, Te Meerman GJ, et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 2012;8:e1002431.
Ikram MA, Brusselle GGO, Murad SD, van Duijn CM, Franco OH, Goedegebure A, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol. 2017;32:807–50.
Steenaard RV, Ligthart S, Stolk L, Peters MJ, van Meurs JB, Uitterlinden AG, et al. Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenetics. 2015;7:54.
Li JH, Liu S, Zhou H, Qu LH, Yang JH, starBasev2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–7.
Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69–80.
Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402.
Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. 2015;116:737–50.
Mirza AH, Kaur S, Brorsson CA, Pociot F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE. 2014;9:e105723.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14.
Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Li H, Lv X. Functional annotation of noncoding variants and prioritization of cancer-associated lncRNAs in lung cancer. Oncol Lett. 2016;12:222–30.
Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–69.
Hayes MG, Urbanek M, Hivert MF, Armstrong LL, Morrison J, Guo C, et al. Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies. Diabetes. 2013;62:3282–91.
Inouye M, Ripatti S, Kettunen J, Lyytikainen LP, Oksala N, Laurila PP, et al. Novel Loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012;8:e1002907.
Liu CT, Raghavan S, Maruthur N, Kabagambe EK, Hong J, Ng MC, et al. Trans-ethnic meta-analysis and functional annotation illuminates the genetic architecture of fasting glucose and insulin. Am J Hum Genet. 2016;99:56–75.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13.
Lipovich L, Brown B, Raghavan S, Wessel J, Meigs J. Long non-coding RNA genes are direct disease causal candidates in human metabolic disorders: insights from the GWAS-transcriptome interface. In: RNA meeting 2015. Madison: University of Wisconsin; 2015.
Manning AK, Lipovich L, Goustin A, Leong A, Morrison AC, Brody J, Liu C, Brown JB, Chen Y, Rotter I, Meigs JB, CHARGE Consortium. The fasting glucose associated PPP1R3B locus maps to LOC157273, a long non-coding RNA which represses PPP1R3B expression in primary human hepatocytes. In: SAC meeting 2016. Boston, MA.
Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ, Hubbard SR. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell. 2005;20:325–33.
Travers ME, Mackay DJ, Dekker Nitert M, Morris AP, Lindgren CM, Berry A, et al. Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes. 2013;62:987–92.
Duchatelet S, Crotti L, Peat RA, Denjoy I, Itoh H, Berthet M, et al. Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in long-QT syndrome. Circ Cardiovasc Genet. 2013;6:354–61.
Olszak-Waskiewicz M, Dziuk M, Kubik L, Kaczanowski R, Kucharczyk K. Novel KCNQ1 mutations in patients after myocardial infarction. Cardiol J. 2008;15:252–60.
Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, et al. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96:693–701.
Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res. 2006;98:1055–63.
Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–102.
Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40:1092–7.
Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014;115:668–77.
Inanobe A, Tsuzuki C, Kurachi Y. An epithelial Ca2+-sensor protein is an alternative to calmodulin to compose functional KCNQ1 channels. Cell Physiol Biochem. 2015;36:1847–61.
Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9.
Korostowski L, Sedlak N, Engel N. The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet. 2012;8:e1002956.
Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, et al. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005;33:2650–60.
Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, et al. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic beta-cell mass by epigenetic modification of Cdkn1c. Proc Natl Acad Sci USA. 2015;112:8332–7.
Korostowski L, Sedlak N, Engel N. The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoSGenet. 2012;8:e1002956.
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89.
Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol. 2006;26:9196–208.
Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, et al. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–21.
Vance KW, Sansom SN, Lee S, Chalei V, Kong L, Cooper SE, et al. The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 2014;33:296–311.
Luo H, Sun S, Li P, Bu D, Cao H, Zhao Y. Comprehensive characterization of 10,571 mouse large intergenic noncoding RNAs from whole transcriptome sequencing. PLoS ONE. 2013;8:e70835.
Potter SS, Branford WW. Evolutionary conservation and tissue-specific processing of Hoxa 11 antisense transcripts. Mamm Genome. 1998;9:799–806.
Chau YM, Pando S, Taylor HS. HOXA11 silencing and endogenous HOXA11 antisense ribonucleic acid in the uterine endometrium. J Clin Endocrinol Metab. 2002;87:2674–80.
Richards EJ, Permuth-Wey J, Li Y, Chen YA, Coppola D, Reid BM, et al. A functional variant in HOXA11-AS, a novel long non-coding RNA, inhibits the oncogenic phenotype of epithelial ovarian cancer. Oncotarget. 2015;6:34745–57.
Parrillo L, Costa V, Raciti GA, Longo M, Spinelli R, Esposito R, et al. Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int J Obes. 2016;40:929–37.
Singh S, Rajput YS, Barui AK, Sharma R, Datta TK. Fat accumulation in differentiated brown adipocytes is linked with expression of Hox genes. Gene Expr Patterns. 2016;20:99–105.
Yu W, Peng W, Jiang H, Sha H, Li J. LncRNA HOXA11-AS promotes proliferation and invasion by targeting miR-124 in human non-small cell lung cancer cells. Tumour Biol. 2017;39:10.
Hartig MB, Iuso A, Haack T, Kmiec T, Jurkiewicz E, Heim K, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2011;89:543–50.
Langenberg C, Sharp SJ, Franks PW, Scott RA, Deloukas P, Forouhi NG, et al. Gene-lifestyle interaction and type 2 diabetes: the EPIC interact case-cohort study. PLoS Med. 2014;11:e1001647.
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90.
Angunsri N, Taura A, Nakagawa T, Hayashi Y, Kitajiri S, Omi E, et al. Insulin-like growth factor 1 protects vestibular hair cells from aminoglycosides. Neuroreport. 2011;22:38–43.
Acknowledgements
Mohsen Ghanbari was supported by grants from the Iranian Ministry of health and Mashhad University of Medical Sciences. Oscar H. Franco works in ErasmusAGE, a center for aging research across the life course funded by Nestlé Nutrition (Nestec Ltd.), Metagenics Inc., and AXA. Nestlé Nutrition (Nestec Ltd.), Metagenics Inc., and AXA were not involved in designing and conducting the study; in collection, management, analysis, and interpretation of the data; and preparation, review, and approval of the manuscript. Caren Smith is supported by the NIH National Heart Lung and Bool Institute award K08 HL112845. Jose Ordovas’s lab is partially supported by contracts 53-K06-5-10 and 58-1950-9-001 from the US Department of Agriculture, Agriculture Research Service. The Rotterdam Study is supported by Erasmus MC (Erasmus Medical Center Rotterdam), the Erasmus University Rotterdam, the Netherlands Organization for Scientific Research (NWO), the Netherlands Organization for Health Research and Development (ZonMW), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, and the Ministry of Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The gene expression and DNA methylation data for the Rotterdam Study were funded by the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by the Netherlands Organization for Scientific Research (NWO; project number 184021007). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors are grateful to the Rotterdam Study participants, the staff involved with the Rotterdam Study, and the participating general practitioners and pharmacists. We are also grateful to the GLGC, GIANT, Global BPGen, CARDIoGRAM plusC4D, and DIAGRAM consortia for sharing the GWAS data. We would like to thank the MuTHER Study for performing the eQTL analysis in adipose tissue.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Marjolein J. Peters and Paul S. de Vries contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ghanbari, M., Peters, M.J., de Vries, P.S. et al. A systematic analysis highlights multiple long non-coding RNAs associated with cardiometabolic disorders. J Hum Genet 63, 431–446 (2018). https://doi.org/10.1038/s10038-017-0403-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0403-x
This article is cited by
-
Lipid Phenotypes and DNA Methylation: a Review of the Literature
Current Atherosclerosis Reports (2021)
-
LncRNA H19 rs4929984 Variant is Associated with Coronary Artery Disease Susceptibility in Han Chinese Female Population
Biochemical Genetics (2021)
-
Objectives, design and main findings until 2020 from the Rotterdam Study
European Journal of Epidemiology (2020)
-
Long noncoding RNAs in lipid metabolism: literature review and conservation analysis across species
BMC Genomics (2019)
-
Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes
Human Genomics (2018)
