
Abstract
Major depressive disorder is a common psychiatric disorder that is thought to be triggered by both genetic and environmental factors. Depressive symptoms are an important public health problem and contribute to vulnerability to major depression. Although a substantial number of genetic and epigenetic studies have been performed to date, the detailed etiology of depression remains unclear and there are no validated biomarkers. DNA methylation is one of the major epigenetic modifications that play diverse roles in the etiology of complex diseases. In this study, we performed an epigenome-wide association study (EWAS) of DNA methylation on subjects with (N = 20) or without (N = 27) depressive symptoms in order to examine whether different levels of DNA methylation were associated with depressive tendencies. Employing methylation-array technology, a total of 363,887 methylation sites across the genomes were investigated and several candidate CpG sites associated with depressive symptoms were identified, especially annotated to genes linked to a G-protein coupled receptor protein signaling pathway. These data provide a strong impetus for validation studies using a larger cohort and support the possibility that G-protein coupled receptor protein signaling pathways are involved in the pathogenesis of depression.
Similar content being viewed by others

Introduction
Major depressive disorder is a common psychiatric disorder linked to both genetic and environmental factors [1, 2]. Depressive symptoms are important public health problems and contribute to vulnerability to major depression, which causes suicidal behavior [3]. According to a WHO report, more than 800,000 people die by suicide every year worldwide [4]. Further, 3–33% of adults were reported to experience suicidal ideation during a lifetime [5], and over 90% of suicide victims or suicide attempters commonly have a mood disorder including depression [6]. Previous trials confirmed that preventive action against suicide, such as early treatment and mental health promotion can reduce the incidence by 22% [7, 8]. Therefore, it is important to identify robust biomarkers for depression that can facilitate detection at an early stage.
Family and twin studies have indicated that genetic and environmental factors are important in the pathogenesis of major depression. Estimates of major depressive disorder heritability range from 31 to 42% [2]. However, neither candidate gene approach studies nor genome-wide association studies (GWASs) have identified genes that are associated with depression and are replicated in multiple studies [9] except for two SNPs located in Kinase Suppressor of Ras 2 (KSR2) and DCC Netrin 1 Receptor (DCC) [10]. This inconsistency is thought to be associated with a lack of power partly because the role of environment has not been taken into account [9]. It is widely believed that environmental factors, such as stress might play an important role in the pathogenesis of depression.
Epigenetics is now generally understood to refer to potentially heritable molecular modifications that regulate gene expression and chromatin structure independently from the primary DNA sequence [11]. DNA methylation is one of the major forms of epigenetic modification that play diverse roles in the etiology of complex diseases [12]. Previous studies have shown that DNA methylation is affected by stressful life events and mediates risk in psychiatric disorders [13]. Methylation of the gene encoding the Glucocorticoid Receptor, NR3C1, which is one of the most investigated genes in depression studies, was reported to be associated with early life stress and stress-related psychiatric disorders [13, 14]. The associations of methylation levels with other candidate genes involved in depression, such as brain derived neurotrophic factor (BDNF) and Serotonin Transporter 1 (SLC6A4), have also been reported [13] and interestingly, epigenetic modifications of these genes were found to correspond to antidepressant medication [15]. However, the reported methylation sites of these genes and the effects of methylation on gene expression were not always consistent between studies [13, 14]. Recent technological advances have facilitated the investigation of DNA methylation status across the entire genome. Several epigenome-wide association studies (EWASs) for depression have been performed, and a number of additional candidate genes were identified. Córdova-Palomera et al. [16] reported that most differentially methylated sites examined in their EWAS study were located in genes related to neuropsychiatric phenotypes. Sabunciyan and his colleagues also reported the association of methylation with neuronal development genes [17]. However, these EWASs did not find the associations between methylation and traditional candidate genes linked to depression, including NR3C1, BDNF, and SLC6A4 [16,17,18,19,20,21]. Further, most of the newly identified candidate methylation loci in EWASs were not replicated in other independent studies [16,17,18,19,20,21], clouding the interpretation of these data.
In this study, we performed an EWAS for depressive symptoms on a small Japanese cohort using the methylation array technology. We investigated the methylation loci related to depressive symptoms in healthy individuals who were all free from the antidepressant medication. According to the previous studies, antidepressant have an influence on the DNA methylation status of several genomic loci [15]. Therefore, analysis with medication-free individuals with or without depressive symptoms may be an effective way to detect DNA methylation sites associated with depression. On this basis, we divided healthy individuals with or without depressive symptoms into two groups according to a depression self-rating scale and compared DNA methylation profiles of the two groups.
Materials and methods
Sample
We recruited healthy unrelated Japanese individuals living in Tokyo. The healthy control subjects were interviewed by psychiatrists and filled out a questionnaire, MINI [22], to exclude history of major psychiatric illnesses. They also completed another self-reported questionnaire, CES-D [23], to examine whether they had depressive symptoms. Then, 47 subjects were selected for the current study from our larger sample set described above. They were divided into two groups, with (CES-D ≥ 16, N = 20) and without (CES-D < 16, N = 27) depressive symptoms matching a male/female ratio in order to investigate methylation sites related to depressive tendencies. All the subjects with CES-D ≥ 16 were not diagnosed as major depressive disorder. The annotation data of these samples are provided in Supplementary Table 1. Written informed consent was obtained from each subject. Ethical approval was obtained from the ethical committees of the University of Tokyo.
Epigenome-wide DNA methylation analysis
The DNA methylation data were collected in our previous EWAS study [24]. Briefly, genomic DNA was extracted from white blood cells by the standard procedures (Wizard genomic DNA purification kit, Promega Corporation, WI). DNA samples were first bisulfite-converted using EZ DNA Methylation Kit (Zymo Research, Irvine, CA). The levels of DNA methylation were examined with a DNA methylation array (Infinium Human Methylation 450 K BeadChip, Illumina Inc., CA) according to the manufacturer’s protocol. The DNA samples were amplified by a whole-genome amplification technique and then fragmented and hybridized to the methylation array. After the hybridization, a single-base extension step determined the DNA methylation status of each locus. The arrays were imaged with a high-precision scanner (iScan system, Illumina Inc.) and the signal intensities corresponding to methylated or unmethylated signals were extracted with a software package (GenomeStudio Software, Illumina Inc.). The DNA methylation status of each cytosine residue was evaluated using β- and M-values. The β-value is the ratio of the signal from the methylated probe to the total signal intensity and ranges from 0 (unmethylated) to 1 (completely methylated). The M-value is the logit transformation of the β-value, which is statistically valid for the differential analysis of methylation levels [25].
Data filtering and normalization
The β-values generated by GenomeStudio Software were subjected to stringent data filtering and normalization. First, β-values with a detection P-value ≥ 0.01 were treated as missing values and the call rate of each probe was calculated. We included probes which met the following conditions in the EWAS analysis: (1) probe call rate > 95%, (2) probe on autosomal chromosome, (3) probe not including a single-nucleotide polymorphism (SNP) with a minor allele frequency ≥ 0.05, and (4) probe not reported to have cross-reactivity [26] (Supplementary Fig. 1). A previous study reported probes that co-hybridize to alternate sequences that are highly homologous (< 4 base mismatches among 50 bases) to the intended targets [26] as cross-reactive probes. In addition to these probes, we excluded the additional potentially cross-reactive probes. We created a list of possible cross-reactive probes that had 20-bp of sequence from the 5′ end that was perfectly matched to unintended target sequences [24]. After filtering, we conducted stringent data normalization using the following pipeline: (1) Lumi: color bias correction and quantile normalization (QN; correction for the distributions of the pooled probes) [27], (2) beta-mixture quantile dilation (BMIQ) normalization (correction for probe design bias) [28], (3) correction for the batch effect (ComBat) [29].
To investigate methylation sites associated with depressive symptoms, the normalized β-values were converted to M-values and applied to the linear regression analysis.
Prediction of the proportions of leukocyte subtypes
To address the effect of different distributions of leukocyte subtypes between subjects with and without depressive symptoms, we predicted the proportions of each leukocyte subtype in all subjects. The proportions of natural killer (NK) cells, B cells, CD4+ T cells, CD8+ T cells, monocytes, and granulocytes were estimated using a published algorithm [30, 31]. The estimated proportions were used to control for the effects of leukocyte subtypes in the linear regression analysis.
Epigenome-wide association analysis
For the EWAS, significant associations were assessed by linear regression analysis with adjustments for the effects of age, sex, BMI, and the predicted proportions of leukocyte subsets. M-values were used for the analysis. In order to rank the differentially methylated positions (DMPs) efficiently, we employed a combined-rank scheme [32]. In EWAS analyses, probes with the lowest P-values do not always have large effects, i.e., statistically significant probes sometimes have very small β-value differences between case and control subjects (delta β; Δβ) and are considered not to be biologically significant [33]. The combined-rank method considers both P-value and Δβ, therefore, we can prioritize DMPs which are important in the context of both statistical significance and effect size. P-values calculated with the linear regression model and adjusted Δβ [(adjusted case β-value) − (adjusted control β-value)] were used to calculate combined-ranks. First we ranked methylation sites in accordance with P-values and secondly the methylation sites were ranked in accordance with Δβ. The summation of the two kinds of ranks was then evaluated and the final combined-ranks of the methylation sites were determined.
Pathway analysis
In order to investigate the characteristics of the top-ranked CpG sites, pathway analyses were performed with MetaCore software (version 6.24 build 67895, Thomson Reuters, New York, NY). Genes annotated to the top 100 ranked CpG sites were tested to examine whether they had any enrichment of gene sets for biological processes or molecular functions in the GO database (http://geneontology.org/) [34]. Among the detected gene sets, those with more than 500 registered genes were ignored because such gene sets tend to represent broader categories with biological meanings that are often ambiguous [35]. Gene sets with < 5 registered genes that might be less noteworthy as “gene sets” were also disregarded. Further, we also conducted another GO-based pathway analysis with GOseq software [36], which enabled us to correct for gene length bias, because larger genes usually have many CpG sites and a higher a-priori chance of being included in the pathway analysis [37]. Pathways identified as significant in both MetaCore and GOseq analyses were regarded as candidate pathways.
Assessment of the blood-brain correlation in the top-ranked probes
The top 100 ranked probes were examined to determine whether the methylation status of these probe sites correlate between blood and brain. We used the Blood Brain DNA Methylation Comparison Tool (http://epigenetics.iop.kcl.ac.uk/bloodbrain/) to investigate the Pearson’s correlation coefficients of β-values between samples obtained from blood and four brain regions; prefrontal cortex (PFC), entorhinal cortex (EC), superior temporal gyrus (STG) and cerebellum (CER) [38]. The correlation plots were also obtained using this tool.
Statistical analysis
In the pathway analysis, the detected pathway was assessed using the significance level at FDR 0.05. All analyses other than MetaCore-based pathway analysis were performed using R software.
Results
Epigenome-wide DNA methylation analysis
Genome-wide DNA methylation profiles were examined in healthy individuals with or without depressive symptoms using array-based technology. First, a quality check of the array data was performed. We investigated the detection P-values of each probe, which reflect the overall probe performance. More than 99% of all probes in all samples had a detection P-value ≤ 0.05, showing that the overall performance of the assay was high. Density plots of the β-values of each sample were checked by a visual inspection. All plots had a standard bimodal distribution of the β-values as we reported previously [24].
After stringent data filtering and normalization were performed, 363,887 methylation sites were analyzed (Supplementary Fig. 1). DMPs associated with depressive symptoms were investigated by linear regression analyses. Since several DNA methylation sites were observed to be affected by sex, age, and BMI as previous studies, we included these variables as covariates. Cell types have also been reported to have a strong influence on DNA methylation at several specific sites; thus, we estimated the proportions of leukocyte subtypes using a published algorithm. No obvious proportion difference between subjects with or without depression were observed in any of the cell subtypes (Supplementary Fig. 2). However, considering their large impact on DNA methylation, the effects of cell subtypes were also regressed out.
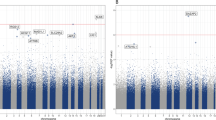
The 100 top-ranked DMPs are listed in (Table 1, Supplementary Table 2). Of the top-ranked DMPs, 85/100 probes (85%) were found to be hypermethylated in the individuals with depressive symptoms, and only 15% were hypomethylated (Fig. 1, Supplementary Table 2). The top-ranked DMP, cg17277199, was 10.7% hypermethylated in subjects with depressive symptoms (P = 9.24 × 10−7, Table 1), and was located in a region within 200-bp upstream of the C2orf84 transcription start site. cg13768055 (RPH3AL, P = 3.27 × 10−4, Δβ = −17.7%), and cg01343041 (C2orf84, P = 8.07 × 10−6, Δβ = 9.6%) were the next two most highly ranked sites.

Result of the EWAS examining DMPs between subjects with and without depressive symptoms. Log-transformed P-values of all the probes were plotted against differences between average adjusted β-values of subjects with and without depressive symptoms. The top 100 ranked probes are shown in blue dots
The top 100 DMPs were found to have no characteristics based on locations related to genes and CpG islands. However, among the top 100 probes, 36% were located in a putative enhancer region. This percentage was relatively high considering that the overall percentage of probes in enhancer regions was 22.9% (Chi-square P = 1.92 × 10−3).
Pathway analysis
Pathway analyses were performed to investigate whether the genes annotated to the top 100 ranked probes were associated with specific biological processes or molecular functions. Sixty-seven genes located near the top 100 DMPs were used for the two kinds of pathway analyses. First, Metacore software, which is based on the expert-curated data sets, was used to identify networks that showed significant associations at a FDR of 5%. Additionally, we performed pathway analysis with GOseq software in order to adjust for gene-length bias. The “regulation of G-protein coupled receptor protein signaling pathway” was significant in both analyses (P = 1.69 × 10−6, Table 2).
Assessment of the blood-brain correlation in the top-ranked probes
As for the top 100 ranked probes, blood and brain correlations of the methylation were examined using the database. Of the top DMPs, 77/100 (77%) showed at least weak (r ≥ 0.2) association with at least one brain region, and six DMPs had a strong correlation (r ≥ 0.7) (Supplementary Table 3). The DMP with the highest correlation was the second top-ranked probe, cg13768055, with r > 0.8 between blood and three brain regions, PFC, EC, and STG (PFC, r = 0.84; EC, r = 0.82; STG, r = 0.84) (Supplementary Fig. 3).
Discussion
To the best of our knowledge this is the first reported EWAS for depressive symptoms in healthy individuals. The major advantage of this study was the investigation of DNA methylation profiles associated with depressive symptoms in subjects who were all free from antidepressant medication. According to a previous study using animal models, selective serotonin reuptake inhibitors induced demethylation in the promoter region of the S100 Calcium Binding Protein A10 (S100A10) gene, for which hypermethylation was reported to be associated with the pathophysiology of depression [39]. Another study using animal models revealed that a tricyclic antidepressant imipramine repressed Serotonin 1A Receptor (5-HT1A) expression through the demethylation of its promoter region which is recognized and repressed by Sp4 [40]. These results suggest that antidepressant medication can change the DNA methylation profiles of CpG sites associated with depression. The participants of this study were all healthy human individuals, therefore, several candidate CpG sites that were related to depressive symptoms might reflect the direct effects of depression, free from any potential distortion associated with treatment medications.
The top-ranked probe associated with depressive symptoms, cg17277199, was located upstream of the transcription start site of the C2orf84 gene, although the association did not achieve genome-wide significance. C2orf84, also known as Family With Sequence Similarity 228 member A (FAM228A) encodes a protein of unknown function. The second top-ranked probe, cg13768055, was located in the 8th exon of the Rabphilin 3A-Like (RPH3AL) gene. This CpG site was reported to be a CTCF binding site and hypomethylation of this site may be related to increased CTCF binding, which might in turn regulate RPH3AL expression. Although the detailed function of RPH3AL in humans is still unclear, the protein was found to regulate calcium-ion-dependent exocytosis in endocrine and exocrine cells [41,42,43].
Among the top 100 ranked probes, 71 probes were annotated to a gene region, and about half of them (35/71) were located upstream (within 1500 bp of the transcription start site, 5′ UTR, and 1st exon) of the annotated genes. All the probes in the upstream regions, other than two, were found to be hypermethylated in the subjects with depressive symptoms. Although recent studies have highlighted complex context-dependent interactions between DNA methylation and gene expression [44], a number of previous studies reported that DNA hypermethylation of gene promoter region is related to a lower level of gene expression [45]. Therefore, DNA hypermethylation in upstream regulatory regions in subjects with depressive symptoms might be associated with lower expression of particular genes. Further, many of the top-ranked probes (36/100) were identified in putative enhancer regions raising the possibility of additional effects on gene expression.
In this study, we examined the DNA methylation profiles associated with depressive symptoms using DNA extracted from peripheral blood. Although the methylation profiles were found to be different between blood and brain, several CpG sites were correlated between the two tissues. We examined whether the methylation of the top-ranked DMPs correlated between blood and four brain regions using a published database [38]. From this analysis, 77/100 (77%) of the top DMPs showed at least weak (r ≥ 0.2) associations with at least one brain region, and six DMPs showed strong correlations (r ≥ 0.7) (Supplementary Table 3). The DMP with the highest correlation was the second top-ranked probe, cg13768055, with r > 0.8 between blood and three brain regions, PFC, EC, and STG. Although, detailed studies using brain samples are essential to find additional and/or brain-specific DMPs associated with depressive symptoms, such DMPs with high correlation in this study may be more likely to be related to depressive symptoms through methylation alterations in brain.
Finally, we performed pathway analyses with the genes annotated to the top-ranked DMPs to investigate the functional relationships between these genes. We found that the pathway, regulation of G-protein coupled receptor protein signaling, was significantly associated with depressive symptoms, reflecting the top-ranked probes annotated to RGS14, RGS18, GNG4, CHGA, RPH3AL, and NPR2. Regulator of G-protein Signaling 14 (RGS14) and Regulator of G-protein Signaling 18 (RGS18) are both members of the regulator of G-protein signaling family and bind GTP-bound G-protein alpha subtypes through their RGS domains, increasing the GTPase activity and attenuating GPCR signaling [46, 47]. Selective expression of RGS family members was previously reported [48]. RGS14 was found to be enriched in hippocampal CA2 neurons [47], whereas RGS18 is predominantly expressed in platelets and granulocytes [46]. G-protein subunit gamma 4 (GNG4) is a component of a heterotrimeric G-protein subunit. G-protein alpha, beta, and gamma subunits have preferential interactions and subunit combinations contribute to the specificity of G-protein-mediated signaling pathways [49]. Interestingly, RGS14, RGS18, and GNG4 are all upstream regulators of the G-protein coupled receptor protein signaling pathway. Accumulating evidence suggests that G-protein coupled receptor related pathways are associated with mood disorders including depression [50, 51]. Monoamine receptors including serotonin, norepinephrine, and dopamine receptors are important targets of antidepressant medication and are all G-protein coupled receptor, with the exception of 5-HT3. Antidepressant drugs are believed to work by activating serotonergic, noradrenergic, and dopaminergic receptors and increasing monoamine transmission in the brain [51]. Although the precise mechanisms linking G-protein coupled receptors to the antidepressant drug response remain unclear, G-protein coupled receptors are predicted to play a role in the induction of hippocampal neurogenesis [51]. These data are consistent with an association between the regulation of G-protein coupled receptor protein signaling pathway and depressive symptoms found in this study.
In the current study, we could not find any DMPs which were ranked in the top 1000 and were overlapped with depression-associated DMPs reported in the previous studies using blood samples and the same array technology [52,53,54,55]. However, the methylation sites located in protocadherin (PCDH) family genes were reported as highly variable methylation sites according to depression in a previous study [16] and in the current study, we also found several CpG sites with Δβ > 5% and P < 0.05 in this region (Supplementary Table 4). Therefore the PCDH family gene region might be an interesting candidate for future studies.
There are several limitations to this work. First of all, the sample size was small, potentially compromising the power of the statistical analyses. In order to identify additional depression-associated methylation sites, future studies with larger numbers of samples are essential. Second, the current study would benefit from replication using other methods such as pyrosequencing to examine the levels of DNA methylation. Finally, we used DNA extracted from peripheral blood in this study. Although many of the top-ranked CpG sites were consistent between blood and brain, it is necessary to investigate DNA methylation profiles using brain samples to find additional brain-specific DMPs.
In conclusion, we performed an EWAS for depressive symptoms and found several candidate CpG sites associated with this symptoms, especially annotated to genes linked to the G-protein coupled receptor protein signaling pathway. These findings provide a strong impetus for further validation studies using larger cohort to confirm and potentially expand on these result.
Data availability
The data sets of the current study are freely available from the National Bioscience Database Center (NBDC) website (http://biosciencedbc.jp/en/).
References
Lohoff FW. Overview of the genetics of major depressive disorder. Curr Psychiatry Rep. 2010;12:539–46.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Yang L, Jia CX, Qin P. Reliability and validity of the Center for Epidemiologic Studies Depression Scale (CES-D) among suicide attempters and comparison residents in rural China. BMC Psychiatry. 2015;15:76.
World Health Organization Preventing suicide: a global imperative. WHO Library Cataloguing-in-Publication Data. 2014. ISBN: 978 92 4 156477 9.
Nock MK, Borges G. Ono Y Suicide: Global Perspectives from the WHO World Mental Health Surveys. Cambridge Medicine. 2012. ISBN-13: 978-0521765008.
Mann JJ. Neurobiology of suicidal behaviour. Nat Rev Neurosci. 2003;4:819–28.
Muñoz RF, Cuijpers P, Smit F, Barrera AZ, Leykin Y. Prevention of major depression. Annu Rev Clin Psychol. 2010;6:181–12.
Aarø LE, Herbeć A, Bjørngaard JH, Mańczuk M, Zatoński WA. Depressive episodes and depressive tendencies among a sample of adults in Kielce, south-eastern Poland. Ann Agric Environ Med. 2011;18:273–78.
Dunn EC, Brown RC, Dai Y, Rosand J, Nugent NR, Amstadter AB, et al. Genetic determinants of depression: recent findings and future directions. Harv Rev Psychiatry. 2015;23:1–18.
Okbay A, Baselmans BM, De Neve JE, Turley P, Nivard MG, Fontana MA, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet. 2016;48:624–33.
Bird A. Perceptions of epigenetics. Nature. 2007;447:396–98.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology. 2014;80:115–32.
Menke A, Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin Neurosci. 2014;16:395–404.
Lisoway AJ, Zai CC, Tiwari AK, Kennedy JL. DNA methylation and clinical response to antidepressant medication in major depressive disorder: A review and recommendations. Neurosci Lett. 2017.
Córdova-Palomera A, Fatjó-Vilas M, Gastó C, Navarro V, Krebs MO, Fañanás L. Genome-wide methylation study on depression: differential methylation and variable methylation in monozygotic twins. Transl Psychiatry. 2015;5:e557.
Sabunciyan S, Aryee MJ, Irizarry RA, Rongione M, Webster MJ, Kaufman WE, et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS ONE. 2012;7:e34451.
Dempster EL, Wong CC, Lester KJ, Burrage J, Gregory AM, Mill J, et al. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry. 2014;76:977–83.
Weder N, Zhang H, Jensen K, Yang BZ, Simen A, Jackowski A, et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J Am Acad Child Adolesc Psychiatry. 2014;53:417–24.e415.
Davies MN, Krause L, Bell JT, Gao F, Ward KJ, Wu H, et al. Hypermethylation in the ZBTB20 gene is associated with major depressive disorder. Genome Biol. 2014;15:R56.
Byrne EM, Carrillo-Roa T, Henders AK, Bowdler L, McRae AF, Heath AC, et al. Monozygotic twins affected with major depressive disorder have greater variance in methylation than their unaffected co-twin. Transl Psychiatry. 2013;3:e269.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:22–33. quiz34–57
Radloff LS. The use of the Center for Epidemiologic Studies Depression Scale in adolescents and young adults. J Youth Adolesc. 1991;20:149–66.
Shimada-Sugimoto M, Otowa T, Miyagawa T, Umekage T, Kawamura Y, Bundo M, et al. Epigenome-wide association study of DNA methylation in panic disorder. Clin Epigenetics. 2017;9:6.
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9.
Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–48.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–96.
Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11:1138–40.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9.
Pan W, Kwak IY, Wei P. A Powerful Pathway-Based Adaptive Test for Genetic Association with Common or Rare Variants. Am J Hum Genet. 2015;97:86–98.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14.
Geeleher P, Hartnett L, Egan LJ, Golden A, Raja Ali RA, Seoighe C. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics. 2013;29:1851–57.
Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–32.
Melas PA, Rogdaki M, Lennartsson A, Björk K, Qi H, Witasp A, et al. Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychopharmacol. 2012;15:669–79.
Le François B, Soo J, Millar AM, Daigle M, Le Guisquet AM, Leman S, et al. Chronic mild stress and antidepressant treatment alter 5-HT1A receptor expression by modifying DNA methylation of a conserved Sp4 site. Neurobiol Dis. 2015;82:332–41.
Kato M, Sasaki T, Ohya T, Nakanishi H, Nishioka H, Imamura M, et al. Physical and functional interaction of rabphilin-3A with alpha-actinin. J Biol Chem. 1996;271:31775–78.
Kotake K, Ozaki N, Mizuta M, Sekiya S, Inagaki N, Seino S. Noc2, a putative zinc finger protein involved in exocytosis in endocrine cells. J Biol Chem. 1997;272:29407–10.
Ogata S, Miki T, Seino S, Tamai S, Kasai H, Nemoto T. A novel function of Noc2 in agonist-induced intracellular Ca2+ increase during zymogen-granule exocytosis in pancreatic acinar cells. PLoS ONE. 2012;7:e37048.
Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37.
Lou S, Lee HM, Qin H, Li JW, Gao Z, Liu X, et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014;15:408.
Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–95.
Vellano CP, Lee SE, Dudek SM, Hepler JR. RGS14 at the interface of hippocampal signaling and synaptic plasticity. Trends Pharmacol Sci. 2011;32:666–74.
Larminie C, Murdock P, Walhin JP, Duckworth M, Blumer KJ, Scheideler MA, et al. Selective expression of regulators of G-protein signaling (RGS) in the human central nervous system. Brain Res Mol Brain Res. 2004;122:24–34.
Ray K, Kunsch C, Bonner LM, Robishaw JD. Isolation of cDNA clones encoding eight different human G protein gamma subunits, including three novel forms designated the gamma 4, gamma 10, and gamma 11 subunits. J Biol Chem. 1995;270:21765–71.
Schreiber G, Avissar S. Regulators of G-protein-coupled receptor-G-protein coupling: antidepressants mechanism of action. Expert Rev Neurother. 2007;7:75–84.
Pavlovicova M, Lacinova L, Dremencov E. Cellular and molecular mechanisms underlying the treatment of depression: focusing on hippocampal G-protein-coupled receptors and voltage-dependent calcium channels. Gen Physiol Biophys. 2015;34:353–66.
Numata S, Ishii K, Tajima A, Iga J, Kinoshita M, Watanabe S, et al. Blood diagnostic biomarkers for major depressive disorder using multiplex DNA methylation profiles: discovery and validation. Epigenetics. 2015;10:135–41.
Walker RM, Christoforou AN, McCartney DL, Morris SW, Kennedy NA, Morten P, et al. DNA methylation in a Scottish family multiply affected by bipolar disorder and major depressive disorder. Clin Epigenetics. 2016;8:5.
Walker RM, Sussmann JE, Whalley HC, Ryan NM, Porteous DJ, McIntosh AM, et al. Preliminary assessment of pre-morbid DNA methylation in individuals at high genetic risk of mood disorders. Bipolar Disord. 2016;18:410–22.
Boström AE, Ciuculete DM, Attwood M, Krattinger R, Nikontovic L, Titova OE, et al. A MIR4646 associated methylation locus is hypomethylated in adolescent depression. J Affect Disord. 2017;220:117–28.
Acknowledgments
We thank all the participants of this study. This study was supported by JSPS (JSPS KAKENHI Grant Number 15J04964, 26461712, and 25461723) and the Takeda Science Foundation in Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Shimada, M., Otowa, T., Miyagawa, T. et al. An epigenome-wide methylation study of healthy individuals with or without depressive symptoms. J Hum Genet 63, 319–326 (2018). https://doi.org/10.1038/s10038-017-0382-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0382-y
This article is cited by
-
Immune targets for therapeutic development in depression: towards precision medicine
Nature Reviews Drug Discovery (2022)
-
DNA hypomethylation of the Krüppel-like factor 11 (KLF11) gene promoter: a putative biomarker of depression comorbidity in panic disorder and of non-anxious depression?
Journal of Neural Transmission (2020)
