
Abstract
Background
The mechanisms underlying aberrant activation of intestinal Toll-like receptor 4 (TLR4) signaling in necrotizing enterocolitis (NEC) remain unclear. In this study, we examined the role of single-immunoglobulin interleukin-1 receptor-related molecule (SIGIRR), an inhibitor of TLR signaling, in modulating experimental NEC vulnerability in mice.
Methods
Experimental NEC was induced in neonatal wild-type and SIGIRR−/− mice using hypoxia, formula-feeding, and lipopolysaccharide administration. Intestinal TLR canonical signaling, inflammation, apoptosis, and severity of experimental NEC were examined at baseline and after NEC induction in mice.
Results
SIGIRR is developmentally regulated in the neonatal intestine with a restricted expression after birth and a gradual increase by day 8. At baseline, breast-fed SIGIRR−/− mouse pups exhibited low-grade inflammation and TLR pathway activation compared with SIGIRR+/+ pups. With experimental NEC, SIGIRR−/− mice had significantly more intestinal interleukin (IL)-1β, KC (mouse homolog to IL-8), intercellular adhesion molecule-1 (ICAM-1), and interferon-beta (IFN-β) expression in association with the amplified TLR pathway activation. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, cleaved caspase 3, and severity of intestinal injury with NEC were worse in SIGIRR−/− mice in comparison with SIGIRR+/+ mice.
Conclusion
SIGIRR is a negative regulator of TLR4 signaling in the developing intestine, and its insufficiency results in native intestinal TLR hyper-responsiveness conducive to the development of severe experimental NEC in mice.
Similar content being viewed by others

Main
Necrotizing enterocolitis (NEC) is a common and devastating intestinal disease that primarily affects premature infants. About 5–14% of infants weighing <1,500 g at birth develop NEC, and about 25–40% of them die because of it(1, 2). Despite the significant advances in neonatal medicine, the prevalence of NEC has not decreased and may even be increasing(3). Many risk factors have been implicated in NEC including prematurity, formula-feeding, abnormal intestinal colonization, hypoxia, infection, and blood transfusion(4). Although the final common pathway underlying NEC remains incompletely understood, current evidence suggests that aberrant activation of the innate immune system, triggered in large part by intestinal dysbiosis, has a central role in its pathogenesis(5).
Toll-like receptors (TLRs) are vital components of the intestinal innate immune system. TLRs function by recognizing unique structural motifs conserved in pathogens and activating downstream transcription factors to mount an effective immune response(6). Under normal conditions, TLR activity is balanced, so that commensal gut bacteria are tolerated, while still providing immune activity against invading pathogens(7). In NEC, there is exaggerated activation of TLR leading to pathologic inflammation, local tissue injury, and a widespread inflammatory response. In particular, Toll-like receptor 4 (TLR4), which is the receptor that recognizes the lipopolysaccharide component of Gram-negative bacteria, has been extensively implicated in NEC(8, 9, 10). Because of the immense capacity of TLR4 to regulate intestinal inflammation, tight regulation of TLR4 signaling is essential to maintain the health of the developing intestinal tract(11). Regulation of intestinal TLR signaling immediately after birth, during the phase of rapid postnatal colonization, remains poorly understood.
Single-immunoglobulin interleukin-1 receptor-related molecule (SIGIRR) is a transmembrane protein widely expressed in the intestinal epithelium. SIGIRR functions as a potent negative regulator of TLR and interleukin-1 receptor (IL-1R)-mediated inflammation in the adult intestine(12, 13). In a pilot study, we used a forward genetic approach to identify rare and novel loss-of-function SIGIRR mutations in premature infants with NEC(14). This discovery, which is the first to report of a disease phenotype associated with SIGIRR in humans, identifies SIGIRR as a promising candidate gene for NEC. Functional studies in intestinal epithelial cells confirmed that a decreased SIGIRR function arising from the identified mutations leads to loss of inhibition of TLR4- and IL-1β-mediated inflammation(14). To understand the pathogenesis of NEC arising from loss of SIGIRR function in the neonatal intestine, we pursued studies in SIGIRR−/− mice using a well-accepted animal model of the disease. We hypothesized that the loss of SIGIRR in the neonatal intestine will increase the vulnerability to TLR-mediated inflammation and intestinal injury. Herein, we demonstrate that SIGIRR−/− mice have native intestinal TLR hyper-responsiveness that translates to exaggerated canonical TLR4 canonical signaling and intestinal injury with the induction of experimental NEC.
Methods
Animal Model
C57BL/6 SIGIRR−/− mice(12) were obtained from Xiaoxia Li and bred at the Medical College of Wisconsin, whereas SIGIRR+/+ mice were commercially obtained from Charles River (Burlington, MA). All studies relating to experimental NEC were performed at Medical College of Wisconsin. Litters of mice were breast-fed by the mother until days of life (DOL) 7–8, separated from the mother, and randomly assigned to either control or NEC groups. NEC was induced using a modified NEC protocol(9). Briefly, the mice were gavage-fed with 0.1 ml every 3 h with a formula (15 g Similac 60/40 in 75 ml Esbilac canine milk replacer) mixed with lipopolysaccharide Escherichia coli 055:B5 (200 μl/5 g of mouse body weight, Sigma, St Louis, MO) five times daily with a 24-Fr angiocatheter. The mice were also exposed to hypoxic stress (5% oxygen and 95% nitrogen) twice daily for 10 min each of exposure. After 4 days of NEC induction, mice were killed on DOL-11–12 using an intraperitoneal injection of a ketamine and xylazine mixture (100 and 10 mg/kg, respectively), and the terminal ileum was harvested. Control pups, which remained with the mother under regular conditions, were killed at DOL-11–12 for comparison with the NEC group.
Additional mice were also bred at the University of Missouri-Kansas City animal facility under normal conditions and killed on DOL-7–8 to compare baseline differences between SIGIRR+/+ and −/− mice before NEC induction. Care of mice was conducted in accordance with the National Institutes of Health guidelines, and all protocols had prior approval from Medical College of Wisconsin and University of Missouri-Kansas City Institutional Animal Care and Use Committee.
Quantification of Inflammatory Cytokine mRNA Expression Using Real-Time PCR
A segment of the terminal ileum was homogenized using the bullet blender (Midwest Scientific, St Louis, MO). Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA), and cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The transcripts were amplified, and gene-expression data were collected on a Bio-Rad 1Q5 with SYBR Green Mastermix. Pre-validated primers for 18S, tumor necrosis factor-alpha (TNF-α), inducible nitric oxide synthase (iNOS), a mouse homolog to IL-8 (KC), interleukin-1 beta (IL-1β), and interferon-beta (IFN-β) were purchased from Sigma. 18S was used as the housekeeping gene. The relative gene expression of TNF-α, iNOS, KC, IL-1β, and IFN-β was calculated with the Pfaffl method(15).
Quantification of the Changes in Protein Expression and Phosphorylation
Clarified lysates from a harvested tissue were used for protein analysis. The samples were lysed in RIPA buffer containing protease and phosphatase inhibitors (Sigma) and were homogenized by the bullet blender (Midwest Scientific). Immunoblotting was done following standard protocols. The primary antibodies for phosphorylation were as follows: rabbit anti-(p)IKK-β(Ser177), rabbit anti-(p)p38 MAPK(Thr180/Tyr182), rabbit anti-(p)SAPK/JNK(Thr183/Tyr185), mouse anti-IKK-β, rabbit anti-p38 MAPK, rabbit anti-SAPK/JNK, rabbit anti-(p)p65(Ser132) (Cell Signaling Technology, Danvers, MA), rabbit anti-p65 (Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-β-Actin (Sigma). Other antibodies used were as follows: mouse anti-intercellular adhesion molecule-1 (ICAM-1), rabbit anti-IL-1β (Santa Cruz Biotechnology), rabbit anti-iNOS (BD Biosciences, San Jose, CA), and rabbit anti-Cleaved Caspase 3(Asp175) (Cell Signaling Technology). Blots were developed using enhanced chemiluminescence and were stripped using Restore Plus stripping buffer (Thermo Fisher, Rockford, IL). β-Actin or the corresponding non-phosphorylated antibody was used for normalization, and densitometry was performed using ImageJ Software (NIH, Bethesda, MD). Whole blots are provided in Supplementary Figures S1 and S2 online.
Immunofluorescence for SIGIRR and TUNEL Assay for Apoptosis
A segment of terminal ileum was used for histology and fixed with neutral-buffered formalin and was embedded in paraffin. The paraffin blocks were cut at 4 μm and adhered to positive-charged slides. The cut sections were subjected to immunofluorescence analysis, as previously described(16). Briefly, the slides were rehydrated, and antigen retrieval was performed with citrate buffer. The slides were washed with phosphate-buffered saline and blocked with 0.1% bovine serum albumin and the corresponding secondary serum. The primary antibody was rabbit anti-SIGIRR (Santa Cruz Biotechnology), and the corresponding Alexa Fluor 488 secondary antibody was used (Life Technologies, Carlsbad, CA). The nucleus was stained with 4,6-diamidino-2-phenylindole (DAPI; Life Technologies). Images were captured using a Nikon Eclipse 90i microscope equipped with a Nikon C-1 Confocal and EZ C1 imaging software. The images were taken at × 20, and processing was performed using ImageJ (NIH).
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed on the terminal ileum slides as per the manufacturer’s instructions (Promega, Madison, WI). The images were captured at × 40 using a Zeiss Observer Z1 fluorescence microscope with an attached camera. Quantitation of the TUNEL assay was done by measuring the total number of DAPI-stained cells (blue) and the total number of the TUNEL-positive cells (green) per high-power field (HPF) using the ImageJ software (NIH), with at least four HPF per sample. The apoptotic index was calculated as the proportion of DAPI+ (blue) cells that were stained for TUNEL+ (green).
Hematoxylin and Eosin Staining to Evaluate the Histologic Grading of NEC
The segment of terminal ileum harvested for histology was used for grading of intestinal injury from NEC. Hematoxylin and eosin staining was performed on 4 μm-thick sections. Two separate investigators blinded to the experimental conditions (D.G. and S.W.) graded tissue samples using a 4-point scale, as previously described(17). The slides were scanned into a computer at × 40 using a Hamamatsu NanoZoomer Slide Scanner, and the analysis was completed using the system’s imaging software (NDP imager; Hamamatsu Photonics, Bridgewater, NJ).
Data Analysis
Data were presented as means±SD. P<0.05 was considered to be significant. A minimum of four or more animals was used for each experimental group. Fold-changes in protein levels were determined relative to the expression in control SIGIRR+/+ mice, and were compared between the groups using ANOVA. For changes in phosphorylation, phosphorylated-to-total protein ratio was calculated for each condition and compared between the groups. For mRNA studies, biological replicates were used, fold-change was calculated relative to the expression in control SIGIRR+/+ mice, and it was compared between the experimental groups using ANOVA. The Bonferroni test was used for multiple comparisons. Statistical analysis was conducted using STATA 11 (StataCorp LP, Dallas, TX).
Results
Intestinal SIGIRR Expression Increases with Postnatal Age in Newborn Mice
Developmental changes in the intestinal innate immune responses may explain why NEC predominantly occurs in premature infants, whereas term infants are largely protected from the disease. As a first step in investigating the role of SIGIRR in NEC, we sought to determine the changes in SIGIRR during normal development of the intestinal tract. Protein expression of SIGIRR was measured in the jejunum and ileum of newborn SIGIRR+/+ mice on DOL-1, 4, 7, and 14. Intestinal expression of SIGIRR was initially low on DOL-1 and gradually increased with postnatal age, achieving peak expression by day 7–8 (Figure 1a–c). Immunofluorescence studies showed that SIGIRR expression was prominent on DOL-7, and localized to the luminal surface of the intestinal villi by DOL-14 (Figure 1e). SIGIRR expression was also developmentally regulated in the rat’s small intestine with a peak expression after DOL-14 (Figure 1d). These data show that SIGIRR expression increases with gut maturation, and its deficiency coincides with the vulnerability of the rodent’s intestine to experimental NEC(18).

Development changes in SIGIRR expression in the rodent intestine. Intestinal homogenates at various postnatal ages were immunoblotted for SIGIRR: (a) mouse ileum, (c) mouse jejenum, and (d) rat ileum. (b) Graphical representation of SIGIRR in mouse ileum using densitometry. Fold-change in SIGIRR is relative to the expression in DOL-1. *P<0.01 (DOL-1 vs. DOL-7); **P<0.01 (DOL-1 vs. DOL-14) where n=3. (e) Immunofluorescence studies on paraffin-embedded sections demonstrating the localization of SIGIRR in postnatal intestine (n≥3). DOL, days of life; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule.
SIGIRR−/− Mice have Spontaneous Intestinal Activation of Pro-inflammatory TLR Signaling
To determine whether the loss of SIGIRR in the developing intestine predisposes to a pro-inflammatory milieu in the absence of experimental injury, we examined the baseline differences in cytokine gene expression and TLR activation in SIGIRR−/− and SIGIRR+/+ mice on DOL-8. We compared the levels of inflammatory cytokines implicated in human NEC between the terminal ileum of wild-type and SIGIRR−/− mice on DOL-8. We chose DOL-8 because SIGIRR expression is abundant in wild-type mice developmentally, and experimental NEC induction commenced on DOL-8. We found that control SIGIRR−/− had a two- to threefold increase in the intestinal expression of inflammatory cytokines TNFα, iNOS, and KC (Figure 2a). IL-1β, a ligand for IL-1R, which is a member of TLR–interleukin superfamily inhibited by SIGIRR; moreover, IFN-β, a cytokine that is activated by myeloid differentiation primary response 88 (MyD88)-independent TLR4 signaling, was also significantly upregulated at the RNA level (Figure 2a) in SIGIRR−/− mice. Intestinal protein expression of ICAM-1 and IL-1β was also increased in control SIGIRR−/− pups when compared with SIGIRR+/+ pups (Figure 2b).

SIGIRR−/− mice have an elevated cytokine expression and increased canonical TLR4 signaling compared with that of SIGIRR+/+ mice at baseline. Ileal homogenates on DOL-8 were used to examine the inflammatory gene and protein expression, as well as the expression of proteins regulated by SIGIRR. (a) TNF-α, iNOS, KC, IL-1β, and IFN-β mRNA expression was quantified by qRT-PCR. Fold-change in SIGIRR−/− is relative to the expression in SIGIRR+/+ mice. §P<0.01 (SIGIRR+/+ Control vs. SIGIRR−/− Control) where n≥5. (b,c) ICAM-1 and IL-1β protein expression was probed by immunoblotting ileal homogenates from control SIGIRR+/+ and SIGIRR−/− mice (b), and densitometric quantification expressed relative to control SIGIRR+/+ mice is shown graphically (c). §P<0.01 (ICAM-1 SIGIRR+/+ Control vs. SIGIRR−/− Control); §§P<0.01 (IL-1β SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥3. (d,e) SIGIRR, TLR4, TLR5, and IL-1R expression was probed by immunoblotting (d), and densitometric quantification expressed relative to SIGIRR+/+ mice is shown graphically (e). §P<0.01 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥3. (f,g) IKK-βSer177 and p65Ser132 phosphorylation were examined by immunoblotting (f), and (g) fold-change in phosphorylation normalized for total protein was compared between control SIGIRR+/+ mice and SIGIRR−/− mice by densitometry. §P<0.05 ((p)IKK-β SIGIRR+/+ Control vs. SIGIRR−/− Control); §§P<0.05 ((p)p65 SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥5. ICAM, intercellular adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; qRT-PCR, quantitative reverse-transcriptase PCR; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule; TNF-α, tumor necrosis factor-alpha; TLR4, Toll-like receptor 4.
To explain the differences in cytokine expression among SIGIRR+/+ and SIGIRR−/− mice, we initially probed the expression of innate immune receptors regulated by SIGIRR in the intestine. TLR4 expression was modestly elevated, whereas IL-1R expression was modestly decreased in SIGIRR−/− mice when compared with SIGIRR+/+ mice, but TLR5 expression (a receptor for flagellin from Gram-negative bacteria) was comparable (Figure 2d). We then examined whether intestinal canonical TLR signaling was differentially activated in SIGIRR+/+ and SIGIRR−/− mice during postnatal adaptation. We found an increased phosphorylation of the inhibitor of NF-κB Kinase Beta (IKK-β) and p65 (a subunit of NF-κB, RelA) in SIGIRR−/− mice compared with that in SIGIRR+/+ mice (Figure 2f). Together, these results suggest that even without induction of any injury, control SIGIRR−/− mice have a state of TLR hyper-responsiveness evident by a modest activation of downstream canonical TLR signaling and inflammation.
SIGIRR−/− Mice Demonstrate Increased Intestinal Inflammation in Comparison with SIGIRR+/+ Mice with Experimental NEC
Loss-of-function mutations of SIGIRR are enriched in premature infants who develop NEC, suggesting that SIGIRR attenuates intestinal inflammation in the developing intestine(14). To further investigate this hypothesis, we compared the intestinal expression of inflammatory cytokines in SIGIRR+/+ and SIGIRR−/− mice following NEC induction. Experimental NEC robustly induced the gene expression of TNF-α, iNOS, and KC in SIGIRR+/+ mice, which was further amplified in SIGIRR−/− (Figure 3a). Induction of IL-1β RNA was 10-fold higher with experimental NEC in SIGIRR−/− mice (Figure 3b); moreover, IFN-β, a gene that is induced by TLR4-mediated activation of TRIF-dependent signaling, was also significantly higher in SIGIRR−/− mice (Figure 3b). Protein expression of inflammatory cytokines, including iNOS, IL-1β, and ICAM-1, was also exaggerated with NEC induction in SIGIRR−/− mice (Figure 3c). Taken together, these data show that in the developing intestine loss of SIGIRR results in exaggerated cytokine responses with experimental NEC.

Intestinal cytokine expression is exaggerated in SIGIRR−/− mice with the induction of NEC. Ileal tissue homogenates obtained on DOL-11 following NEC induction were used to assess the inflammatory gene and protein expression. (a) TNF-α, iNOS, KC, and mRNA expression was quantified in ileal homogenates using qRT-PCR. Fold-changes in gene expression are relative to the expression in control SIGIRR+/+ pups on day 11. *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC); §P<0.05 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥4. (b) Changes in IL-1β and IFN-β RNA expression in SIGIRR+/+ and SIGIRR−/− mice. *P<0.05 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.05 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC); §P<0.05 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥4. (c,d) ICAM-1, iNOS, and IL-1β protein expression was probed by immunoblotting ileal homogenates from mice with and without NEC induction (c), and densitometric quantification expressed relative to control SIGIRR+/+ mice is shown graphically (d). *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC); §P<0.05 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥4. ICAM, intercellular adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; NEC, necrotizing enterocolitis qRT-PCR, quantitative reverse-transcriptase PCR; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule; TNF-α, tumor necrosis factor-alpha;.
Canonical TLR4 Pathway Signaling is Exaggerated in SIGIRR−/− Mice with NEC
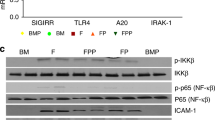
Next, we investigated whether dysregulation of canonical TLR4 signaling in the intestine contributed to an exaggerated inflammatory cytokine expression in SIGIRR−/− mice. We found that NEC-induced IKK-β phosphorylation observed in the intestines of SIGIRR+/+ mice was exaggerated in SIGIRR−/− mice, indicating the activation of NF-κB through a canonical TLR-signaling pathway (Figure 4a). SIGIRR−/− mice with NEC also exhibited an increased phosphorylation of p38 and JNK compared with wild-type mice, indicating an exaggerated TLR4-dependent activation of MAP kinase (MAPK) signaling (Figure 4a). We also examined p65 phosphorylation to determine whether upstream changes in IKK-β phosphorylation and MAPK phosphorylation resulted in NF-κB activation. We found that p65 phosphorylation induced with experimental NEC in SIGIRR+/+ mice was amplified in SIGIRR−/− mice (Figure 4c). Similar to our data on DOL-8 mice, control SIGIRR−/− mice also demonstrated an increased baseline cytokine expression, TLR pathway kinase phosphorylation, and NF-κB activation compared with those in SIGIRR+/+ mice at DOL-11. These results indicate that the insufficiency of SIGIRR results in an exaggerated canonical TLR4 signaling and NF-κB activation that contributes to excessive inflammation seen in SIGIRR−/− mice with experimental NEC.

Activation of canonical TLR4 signaling with NEC induction. Ileal tissue homogenates obtained on day 11 were used to investigate the activation of TLR signaling. (a,b) IKK-βSer177, p38Thr180/Tyr182, and JNKThr183/Tyr185 phosphorylation were examined in ileal homogenates by immunoblotting (a), and fold-change in phosphorylation normalized for the respective protein was compared with control SIGIRR+/+ mice by densitometry (b). *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC); §P<0.05 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥4. (c,d) p65 (NF-κB subunit) phosphorylation was examined in ileal homogenates (c), and fold-change in phosphorylation normalized for the total protein was compared with control SIGIRR+/+ mice (d). *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC); §P<0.05 (SIGIRR+/+ Control vs. SIGIRR−/− Control). n≥4. NEC, necrotizing enterocolitis; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule; TLR4, Toll-like receptor 4.
SIGIRR−/− Mice Demonstrate Increased Intestinal Apoptosis with NEC
Because cell death is a hallmark pathological feature of NEC in infants, we investigated whether SIGIRR−/− mice have increased intestinal apoptosis with NEC. We used TUNEL assay and cleaved caspase-3 levels to measure the degree of apoptosis of enterocytes in SIGIRR−/− and wild-type mice with and without NEC on DOL-11 after completion of the NEC protocol. NEC induced a significant apoptosis in SIGIRR+/+ mice, which was exaggerated by more than twofold in SIGIRR−/− mice (Figure 5a). We noted a mild elevation in cleaved caspase-3 levels in control SIGIRR−/− when compared with that in SIGIRR control pups on DOL-11 by immunoblotting. With experimental NEC, there was further evidence of exaggerated activation of apoptotic signaling in SIGIRR−/− mice in comparison with SIGIRR+/+ mice (Figure 5c). These results indicate that SIGIRR−/− mice exhibit more intestinal injury with experimental NEC than their wild-type counterparts.

Intestinal apoptosis is exaggerated with NEC in SIGIRR−/− mice compared with SIGIRR+/+ mice. (a,b) Representative section from SIGIRR+/+ and SIGIRR−/− mice showing TUNEL+ cells (green) in a DAPI (nuclear stain) background (a), and quantification of an apoptotic index (apoptotic cells/total cells) in intestinal sections (b). *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC). n≥4. (c,d) CC3 expression in SIGIRR+/+ and SIGIRR−/− mice was examined by western-blotting ileal homogenates (c), and quantified using densitometry (d). *P<0.01 (SIGIRR+/+ Control vs. SIGIRR+/+ NEC); **P<0.01 (SIGIRR+/+ NEC vs. SIGIRR−/− NEC). n≥3. CC3, cleaved caspase-3; DAPI, 4,6-diamidino-2-phenylindole; NEC, necrotizing enterocolitis; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.
SIGIRR−/− Mice have More Intestinal Injury Consistent with Experimental NEC
We performed histologic analysis using published scoring criteria to compare intestinal injury in SIGIRR−/− and SIGIRR+/+ mice after NEC induction. There was no difference in NEC injury scores in control animals (Figure 6a), but there were significant differences in injury after experimental NEC. Half of the SIGIRR−/− mice developed grade II+ intestinal injury (NEC grade 1.1 vs. 1.7; P=0.03, n=9) in comparison with SIGIRR+/+ mice, which developed mostly grade-1 NEC (Figure 6a). These results indicate that SIGIRR−/− mice are much more susceptible to experimental NEC as compared with their wild-type counterparts.

Grading of intestinal injury in SIGIRR+/+ and SIGIRR−/− mice after NEC protocol. (a) Representative sections from SIGIRR+/+ and SIGIRR−/− mice on day 11 from control and NEC-induced pups are shown. (b) Graphical representation of intestinal injury. *P<0.01 (SIGIRR+/+ Control (n=7) vs. SIGIRR+/+ NEC (n=9)); **P=0.03 ((SIGIRR+/+ NEC (n=9) vs. SIGIRR−/− NEC (n≥8)). (c) Illustration depicting how SIGIRR deficiency results in a state of intestinal TLR hyper-responsiveness that can induce mild inflammation at baseline conditions, and severe inflammation, apoptosis, and NEC upon exposure to additional stressors. NEC, necrotizing enterocolitis; SIGIRR, single-immunoglobulin interleukin-1 receptor-related molecule; TLR, Toll-like receptor.
Discussion
Human and animal studies strongly suggest that activation of intestinal TLR4 signaling contributes to the pathogenesis of NEC. However, the mechanisms by which aberrant TLR4 signaling is mitigated in the developing intestine are not fully understood. In this study, we evaluated the role of SIGIRR in regulating intestinal TLR4 signaling using a murine model of NEC. We demonstrate that newborn mice deficient in SIGIRR exhibited increased levels of IKK-β phosphorylation and MAPK activation following experimental NEC, consistent with exaggerated activation of canonical TLR4 pathway signaling. SIGIRR−/− mice with NEC had significantly increased intestinal levels of inflammation and apoptosis, as well as more severe NEC compared with wild-type mice. Interestingly, we noted an increased inflammatory cytokine expression in the intestinal tract of SIGIRR-deficient mice under physiologic breast-fed conditions, suggesting an aberrant TLR pathway activation. These data reveal that SIGIRR is an important regulator of TLR4 signaling in the neonatal intestine, and SIGIRR deficiency programs TLR4 hyper-responsiveness conducive to the development of NEC with formula-feeding and hypoxic stress (Figure 6c). Although we used a well-accepted model of NEC, the relevance of SIGIRR to Paneth cell ablation(19) and trinitrobenzene sulfonic-acid models(20) of experimental NEC need to be investigated, as these models probe different pathogenic mechanisms underlying NEC.
Developmental regulation of intestinal innate immune responses may explain why the premature intestine is predisposed to NEC. We thus sought to determine how SIGIRR, a key negative regulator of intestinal TLR4 signaling, is developmentally regulated. We observed that the expression of SIGIRR in the mouse intestine is initially low at birth and increases gradually, peaking around postnatal day 7–10. This pattern of developmental expression, which was also observed in the rat intestine, correlates with previous studies in mice, demonstrating a high intestinal expression of TLR4 at birth that gradually decreases with age(21, 22). The gradual shift from low SIGIRR–high TLR4 expression to high SIGIRR–low TLR4 expression in the developing intestines may explain why term infants and more mature (14–21-day old) mice are protected from developing NEC(18). Our evaluation is limited by a lack of human intestinal samples to evaluate SIGIRR expression in infants, although a similar ontogeny observed in both mouse and rat intestines suggests that the developmental expression of SIGIRR is likely conserved across species. Interestingly, Nanthakumar et al. found a low expression of SIGIRR and other negative regulators of TLR signaling in infants with NEC(23). It is unclear from the above study whether a low SIGIRR expression contributed to the development of NEC or was a response to inflammation in NEC. We speculate based on our data that a low expression of SIGIRR at birth can contribute to the vulnerability of an immature intestine to NEC.
Exaggerated inflammation in the intestinal tract is the main hallmark that characterizes NEC(5). In our study, we observed that breast-fed SIGIRR−/− mice exhibited a modest increase in baseline intestinal inflammation without the induction of experimental injury. Other studies have also noted a mild increase in intestinal inflammation in newborn wild-type mice that is self-limited and attributed to a transient TLR4 pathway activation that occurs postnatally(22). However, our results indicate that a postnatal increase in intestinal inflammation is exaggerated in SIGIRR−/− mice. Xiao et al. (24) also reported a significantly higher expression of inflammatory cytokines in the adult colon of SIGIRR−/− mice compared with that of wild-type mice, suggesting that an exaggerated intestinal inflammation in SIGIRR-deficient states extends to the colon and may not be limited to the immediate newborn period. This exaggerated inflammation in SIGIRR-deficient states supports the hypothesis that the underlying genetic factors predispose certain infants to develop NEC(14, 25). On the basis of our findings and others, we speculate that intestinal loss of SIGIRR, arising from genetic mutations, can prime a state of intestinal TLR hypersensitivity that, in the presence of secondary insults such as formula-feeding and/or dysbiosis, can combine to cause NEC in genetically predisposed preterm infants.
To further evaluate the role of SIGIRR in mitigating NEC, we compared intestinal inflammation in SIGIRR−/− and wild-type mice subjected to a well-established protocol of experimental NEC in newborn mice(18, 26). We observed a significantly elevated expression of inflammatory cytokines in SIGIRR−/− mice with NEC compared with that of wild-type mice. Our previous study also showed a robust expression of inflammatory cytokines, following lipopolysaccharide administration in intestinal epithelial cells expressing loss-of-function SIGIRR mutations(14). Studies by Garlanda et al. (27) and Xiao et al. (24), which examined the role of SIGIRR in relation to inflammatory bowel disease, reported a similar pattern of exaggerated intestinal inflammation in SIGIRR-deficient adult mice treated with dextran sulfate sodium to induce colitis. Taken together, these studies indicate that SIGIRR is important in regulating inflammation in the intestines, and that deficiency in SIGIRR function results in an intestinal phenotype characterized by exaggerated TLR-mediated inflammation.
Among the inflammatory cytokines evaluated in our study, IL-1β was found to be particularly upregulated (by more than 10-fold) in SIGIRR-deficient mice with NEC compared with wild-type mice. IL-1β, which is a potent cytokine that can acutely upregulate inflammation, is consistently elevated in both humans and mice with NEC(28, 29, 30, 31). Increased levels of IL-1β precede the histologic evidence of intestinal injury in NEC, suggesting that IL-1β has an important role in both initiation and propagation of inflammation in NEC(28, 32). A robust upregulation of IL-1β and other cytokines in SIGIRR−/− mice with NEC can be attributed to de-repression of TLR signaling with SIGIRR insufficiency, resulting in increased NF-κβ activation and IL-1β expression. Interestingly, as IL-1β is a ligand for IL-1R, and SIGIRR inhibits IL-1R signaling, IL-1β can upregulate the expression of cytokines, including itself in a NF-κB-dependent manner(13, 33, 34). Together with the above studies, our findings suggest that a combined loss in the ability to dampen the signaling from both TLR4 and IL-1R pathways in SIGIRR-deficient mice can contribute to a vicious cycle of inflammation and intestinal injury perpetuated by 1L-1β and other inflammatory cytokines. Further studies are needed to determine whether blocking IL-1R could mitigate intestinal inflammation and have an efficacy in NEC treatment.
Jilling et al. (8) and Leaphart et al. (9) previously established the importance of TLR4 in NEC. To determine the impact of SIGIRR deficiency in TLR4 signaling, we measured MyD88-dependent signaling in SIGIRR−/− and SIGIRR+/+ mice with and without NEC. We focused on the MYD88-dependent pathway because it is the predominant pathway by which TLR4 signals in NEC(23, 35). Similar to these studies, we found that induction of NEC resulted in increased activation of the MyD88-dependent TLR4 pathway. Furthermore, we found that SIGIRR−/− mice had a significantly increased activation of TLR4 and more severe NEC compared with that of SIGIRR+/+ mice, suggesting that an increased severity of NEC in SIGIRR−/− mice is due to an exaggerated activation of a canonical TLR4-signaling pathway that results from the inability of SIGIRR to inhibit TLR4 activity. Interestingly, IFN-β gene expression was also increased in NEC, and was exaggerated in SIGIRR−/− mice. It is unclear whether the increase in IFN-β expression in our study was related to an underlying SIGIRR deficiency or was a compensatory response (IFN-β has anti-inflammatory roles) to the exaggerated inflammation in SIGIRR−/− mice. The potential role of SIGIRR in regulating IFN-β expression through TRIF in experimental NEC will be investigated in future studies(36).
TLR4-mediated enterocyte apoptosis also has an early and important role in NEC. Rodent studies demonstrate that enterocyte apoptosis precedes tissue injury in NEC(11, 37), and inhibiting apoptosis through an epidermal growth factor or caspase inhibitors reduced the severity of NEC injury(37, 38). Consistent with other studies(9, 35), we observed that intestinal apoptosis is increased, following experimental NEC in association with TLR4 pathway activation. In addition, SIGIRR−/− mice exhibited more severe apoptosis and NEC compared with wild-type mice, suggesting that SIGIRR contributes to TLR4-mediated apoptosis in NEC. Interestingly, Xiao et al. demonstrated that intestinal apoptosis is actually decreased in the colonic epithelium of SIGIRR−/− mice. Their study, however, differs from ours in that enterocyte apoptosis was measured in the colon vs. the small intestine, using SIGIRR−/− adult mice instead of neonatal mice, and following treatment with dextran sulfate sodium to induce intestinal colitis instead of NEC. Previous studies have shown that, although TLR4 activation results in intestinal injury and NEC in the small intestine, TLR4 is also required for optimal enterocyte proliferation in the colon and is protective against colitis(39, 40). Taken in conjunction with our data, these studies suggest that TLR4 activation may have differential effects on apoptosis in different parts of the intestinal tract, during various stages of development, and in different diseases.
In summary, our study provides further evidence for the important role of SIGIRR in regulating TLR4-mediated inflammation in the intestine. Mice deficient in SIGIRR have excessive intestinal inflammation at baseline compared with that of wild-type mice. When exposed to external stressors, SIGIRR−/− mice exhibited unregulated intestinal TLR4 activation, resulting in enhanced intestinal inflammation and increased enterocyte apoptosis. Taken in conjunction with recent studies showing the enrichment of SIGIRR mutation in infants with NEC, our data show that SIGIRR is a critical regulator of TLR signaling in the developing intestine. Future studies will investigate the role of the microbiome in priming TLR sensitivity in SIGIRR−/− mice, and the mechanisms by which SIGIRR regulates TLR and IL-1R signaling in the gut.
References
Neu J, Walker WA . Necrotizing enterocolitis. N Engl J Med 2011;364:255–64.
Fanaroff AA, Stoll BJ, Wright LL et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol 2007;196:147 e141–8.
Patel RM, Kandefer S, Walsh MC et al. Causes and timing of death in extremely premature infants from 2000 through 2011. N Engl J Med 2015;372:331–40.
Ahle M, Drott P, Andersson RE . Epidemiology and trends of necrotizing enterocolitis in Sweden: 1987-2009. Pediatrics 2013;132:e443–51.
Neu J, Pammi M . Pathogenesis of NEC: impact of an altered intestinal microbiome. Semin Perinatol 2017;41:29–35.
Kawai T, Akira S . The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010;11:373–84.
Abreu MT . Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 2010;10:131–44.
Jilling T, Simon D, Lu J et al. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol 2006;177:3273–82.
Leaphart CL, Cavallo J, Gribar SC et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol 2007;179:4808–20.
Afrazi A, Sodhi CP, Richardson W et al. New insights into the pathogenesis and treatment of necrotizing enterocolitis: Toll-like receptors and beyond. Pediatr Res 2011;69:183–8.
Sodhi CP, Neal MD, Siggers R et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012;143:708–18 e701-705.
Wald D, Qin J, Zhao Z et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol 2003;4:920–7.
Qin J, Qian Y, Yao J, Grace C, Li X . SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem 2005;280:25233–41.
Sampath V, Menden H, Helbling D et al. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics 2015;135:e1530–4.
Pfaffl MW . A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29:e45.
Menden HL, Xia S, Mabry SM, Navarro A, Nyp MF, Sampath V . Nicotinamide adenine dinucleotide phosphate oxidase 2 regulates LPS-induced inflammation and alveolar remodeling in the developing lung. Am J Respir Cell Mol Biol 2016;55:767–78.
Whitehouse JS, Xu H, Shi Y et al. Mesenteric nitric oxide and superoxide production in experimental necrotizing enterocolitis. J Surg Res 2010;161:1–8.
Sodhi C, Richardson W, Gribar S, Hackam DJ . The development of animal models for the study of necrotizing enterocolitis. Dis Model Mech 2008;1:94–8.
White JR, Gong H, Pope B, Schlievert P, McElroy SJ . Paneth-cell-disruption-induced necrotizing enterocolitis in mice requires live bacteria and occurs independently of TLR4 signaling. Dis Model Mech 2017;10:727–36.
MohanKumar K, Namachivayam K, Cheng F et al. Trinitrobenzene sulfonic acid-induced intestinal injury in neonatal mice activates transcriptional networks similar to those seen in human necrotizing enterocolitis. Pediatr Res 2017;81:99–112.
Gribar SC, Sodhi CP, Richardson WM et al. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol 2009;182:636–46.
Meng D, Zhu W, Shi HN et al. Toll-like receptor-4 in human and mouse colonic epithelium is developmentally regulated: a possible role in necrotizing enterocolitis. Pediatr Res 2015;77:416–24.
Nanthakumar N, Meng D, Goldstein AM et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS ONE 2011;6:e17776.
Xiao H, Gulen MF, Qin J et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007;26:461–75.
Cuna A, Sampath V . Genetic alterations in necrotizing enterocolitis. Semin Perinatol 2017;41:61–9.
Tanner SM, Berryhill TF, Ellenburg JL et al. Pathogenesis of necrotizing enterocolitis: modeling the innate immune response. Am J Pathol 2015;185:4–16.
Garlanda C, Riva F, Polentarutti N et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci USA 2004;101:3522–6.
Liu Y, Zhu L, Fatheree NY et al. Changes in intestinal Toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2009;297:G442–50.
Yurttutan S, Ozdemir R, Canpolat FE et al. Protective effects of colchicine in an experimental model of necrotizing enterocolitis in neonatal rats. J Surg Res 2013;183:156–62.
Weitkamp JH, Koyama T, Rock MT et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut 2013;62:73–82.
Viscardi RM, Lyon NH, Sun CC, Hebel JR, Hasday JD . Inflammatory cytokine mRNAs in surgical specimens of necrotizing enterocolitis and normal newborn intestine. Pediatr Pathol Lab Med 1997;17:547–59.
Cho SX, Berger PJ, Nold-Petry CA, Nold MF . The immunological landscape in necrotising enterocolitis. Expert Rev Mol Med 2016;18:e12.
Riva F, Bonavita E, Barbati E, Muzio M, Mantovani A, Garlanda C . TIR8/SIGIRR is an interleukin-1 receptor/Toll like receptor family member with regulatory functions in inflammation and immunity. Front Immunol 2012;3:322.
Garlanda C, Anders HJ, Mantovani A . TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol 2009;30:439–46.
Yang G, Bao P, Zhang L et al. Critical role of myeloid differentiation factor 88 in necrotizing enterocolitis. Pediatr Res 2014;75:707–15.
Kawasaki T, Kawai T . Toll-like receptor signaling pathways. Front Immunol 2014;5:461.
Jilling T, Lu J, Jackson M, Caplan MS . Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res 2004;55:622–9.
Clark JA, Lane RH, Maclennan NK et al. Epidermal growth factor reduces intestinal apoptosis in an experimental model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2005;288:G755–62.
Fukata M, Michelsen KS, Eri R et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 2005;288:G1055–65.
Zheng L, Riehl TE, Stenson WF . Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology 2009;137:2041–51.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of Financial Support
This study was supported by institutional funds at Children’s Mercy Hospital and Children’s Hospital of Wisconsin (V.S.), and by the Kenneth L. Smith and Eva S. Smith Clinical Scholar Award (A.C. and V.S.).
Supplementary Information accompanies the paper on the Journal of Perinatology website
Supplementary information
Rights and permissions
About this article

Cite this article
Fawley, J., Cuna, A., Menden, H. et al. Single-Immunoglobulin Interleukin-1-Related Receptor regulates vulnerability to TLR4-mediated necrotizing enterocolitis in a mouse model. Pediatr Res 83, 164–174 (2018). https://doi.org/10.1038/pr.2017.211
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.211
This article is cited by
-
Maternal aryl hydrocarbon receptor activation protects newborns against necrotizing enterocolitis
Nature Communications (2021)
-
Hope on the horizon: promising novel therapies for necrotizing enterocolitis
Pediatric Research (2020)
-
NEC-like intestinal injury is ameliorated by Lactobacillus rhamnosus GG in parallel with SIGIRR and A20 induction in neonatal mice
Pediatric Research (2020)
-
Characterization of the pathoimmunology of necrotizing enterocolitis reveals novel therapeutic opportunities
Nature Communications (2020)
