
Abstract
Background:
Primary distal renal tubular acidosis (DRTA) is a rare disease caused by loss-of-function mutations in at least three genes (ATP6V0A4, ATP6V1B1, and SLC4A1) involved in urinary distal acidification. The next-generation sequencing (NGS) technique facilitates the search for mutations in DRTA patients and helps to characterize the genetic and clinical spectrum of the disease.
Methods:
Ten DRTA patients were studied. They had normal serum anion gap (AG), metabolic acidosis with simultaneous positive urinary AG, and inability to maximally acidify the urine. The exons of the three genes were sequenced in two pools by ultrasequencing. Putative mutations were confirmed by corresponding Sanger sequencing of each exon.
Results:
We found 13 mutations in nine patients. ATP6V0A4: Intron16+2insA; p.R807Q; p.Q276fs; p.P395fs; Intron7-2T>C. ATP6V1B1: p.I386fs; p.R394Q. SLC4A1: p.V245M; p.R589C; p.R589H; p.G609A. One case was a compound heterozygous with a known mutation in ATP6V1B1 (p.G609R) and a pathogenic variation at SLC4A1 (p.E508K). One patient was negative for mutations.
Conclusion:
This study evidences that NGS is labor and cost effective for the analysis of DRTA genes. Our results show for the first time SLC4A1 gene mutations in Spanish patients and disclose that compound heterozygosity at two different genes can be responsible for DRTA.
Similar content being viewed by others


Main
Primary distal renal tubular acidosis (DRTA) (1) is caused by a genetic defect that impairs the ability of kidneys to maximally acidify the urine in the presence of normal serum anion gap hyperchloremic metabolic acidosis. Autosomal-recessive defects in the H+ ATPase activity cause most of the primary DRTA cases. H+ ATPase is a highly conserved proton pump formed by two domains, V1 and V0. The B1 isoform, coded by the ATP6V1B1 gene, forms part of the V1 domain that captures protons from cell cytoplasm. This isoform is expressed by alpha intercalated cells of distal tubule as well as by inner ear and endolymphatic sac cells, and this explains why mutations in this gene are related to DRTA with early-onset sensorineural hearing loss (SNHL) (2). The A4 subunit, coded by the ATP6V0A4 gene, is part of the V0 transmembrane domain involved in proton translocation through the cellular membrane. Mutations in this gene were first related to DRTA with no deafness or late-onset SNHL (3), although it is currently established that deafness cannot discriminate between A4 and B1 subunits’ defects (4). Few cases have been related with defects in the human AE1 (HCO3-/Clex changer) channel, expressed not only by alpha intercalated cells but also by erythrocytes. The AE1 kidney isoform (kAE1) has the same amino acid sequence as the red cell isoform (eAE1), but the N-terminal end is truncated. Although both kAE1 and eAE1 isoforms are coded by the SLC4A1 gene, they have different promoter regions and alternative splicing, which regulate the expression and sequence characteristics of the kidney and erythrocyte isoforms. Thus, mutations in the SLC4A1 gene cause either DRTA and/or hemolytic anemia with red cell morphology anomalies (5). The inheritance of this type of DRTA is complex with both autosomal-dominant (6) and autosomal-recessive (5) transmissions. Approximately 94 mutations (Human Gene Mutation Database) in ATP6V0A4, ATP6V1B1, or SLC4A1 genes have been found to cause DRTA. The identification of these mutations has facilitated a better understanding of the molecular defects underlying DRTA with regard to the alterations in the structure (2,3,5,6,7) and intracellular traffic (4,8) of the involved transporters and channels. To date, the mutational screening of these genes relied in the amplification and Sanger sequencing of the 58 coding exons, what represents a huge effort in technical labor and cost. Next-generation sequencing (NGS) would facilitate the rapid and cost-saving search for mutations in these complex diseases (9). The Ion Torrent Personal Genome Machine (PGM) is a semiconductor NGS technology that has been proved useful to characterize the mutational spectrum of several Mendelian disorders (10,11). In this study, we designated and validated a semiconductor chip-based NGS of the ATP6V0A4, ATP6V1B1, and SLC4A1 genes on 10 patients with primary DRTA enrolled in RenalTube, a collaborative international effort aimed to the study of primary tubulopathies (12).
Results
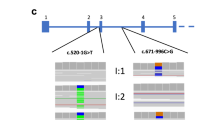
Clinical and biochemical features of the 10 DRTA patients are shown in Table 1 . Patients presented metabolic acidosis (blood pH: 7.19 ± 0.09; serum bicarbonate: 14.1 ± 3.0 mEq/l) with simultaneous positive urinary anion gap and inability to maximally acidify the urine (urine pH 7.03 ± 0.30). Common findings were hypokalemia (serum potassium: 2.9 ± 0.7 mEq/l) and nephrocalcinosis. Three cases were associated to SNHL. Consanguinity was reported in patients 311 and 600 ( Table 1 ). Control cases harboring a known mutation identified by conventional Sanger sequencing (patients 311, 414, and 600) were Hindu, Hispanic, and African, respectively. The rest of cases were Caucasian. The main data of the Ion PGM run is summarized in Supplementary Table S2 online. Only 3 of the 67 amplicons gave poor or null reads in both pools, corresponding to exons 11, 10, and 16 of ATP6V0A4, ATP6V1B1, and SLC4A1, respectively (Supplementary Figure S1 online). Thus, these exons should be Sanger sequenced in all patients as part of mutation screening. We detected all the known control variants in both pools ( Table 2 ). Besides, we identified nine variants that fulfilled our criteria of putative mutations, and all of them were confirmed by Sanger sequencing ( Table 2 ; Supplementary Figure S2 online). NGS and posterior corroboration by Sanger sequencing revealed mutations in nine cases ( Table 2 ). Patients 122 and 323 harbor a missense change in SLC4A1 hotspot p.589 position (13,14) (R589C and R589H, respectively). Neither of their parents are mutation carriers, suggesting a de novo origin in both cases. Patient 122 was also confirmed to carry the SLC4A1 V245M variation inherited from the father. As the father did not have clinical or biochemical manifestations of DRTA, we concluded that it was a polymorphism. Patient 493 was heterozygous for a single described mutation G609R in the SLC4A1 gene (15). He have familial background of DRTA (Renaltube database), his affected mother who is carrier of the same mutation. Case 158 disclosed a homozygosis deletion of a G nucleotide in ATP6V0A4 gene, causing the frameshift change p.P395fs. In patient 786, we confirmed two mutations affecting splicing of the ATP6V0A4 gene mRNA, Intron 16+2 insA (2) and Intron 7−2 T>C (nondescribed). As the variation Intron 7−2 T>C has not been previously described, we corroborated pathogenicity by the Alternative Splice Site Predictor software, which revealed the base involved in a constitutive acceptor zone with a splice site strength score of 2.391 vs. 7.730 with the wild-type base. Patient 306 was carrying the mutation R394Q at the ATP6V1B1 gene (16) and also the variation E508K at the SLC4A1 gene with a predictive deleterious effect according to the PolyPhen software (value = 1) and of not safety by SIFT software (value = 0). To find out the way of segregation of both changes, we analyzed her parents who did not have the ATP6V1B1 gene mutation. The E508K mutation in the SLC4A1 gene was carried by her mother who had not presented DRTA symptoms during her life. Finally, case 491 was the only one without mutations in none of the three analyzed genes.
Discussion
The above results suggest that the NGS platform is a rapid and useful tool for genetic diagnosis. A growing number of publications are reporting this technique for the identification of disease-causing mutations in some Mendelian disorders (9,10,11,17). However, its elevated cost still supposes a drawback. Our study presents several technical features that should be emphasized. We amplified the target amplicons of ATP6V0A4, ATP6V1B1, and SLC4A1 genes and validated the coverage and accuracy of detecting rare variants in two pools of five patients each. This strategy saves the elevated costs of single sample barcoding normally used in NGS experiments. In terms of limitations, it is of note the restrictions related with the Ampliseq design. It is difficult to find probes for all amplicons of several genes because of the presence of regions in genomic DNA with high rates of polymorphisms or guanine/cytosine content. However, in our study focused in the three DRTA genes, we obtained a 96% overall coverage, and only exons 11, 10, and 16 of ATP6V0A4, ATP6V1B1, and SLC4A1, respectively, had to be Sanger sequenced (Supplementary Table S1 online; Supplementary Figure S1 online). Thus, we show, in spite of the limitations resulting from the technique and the pooling methodology, our NGS procedure is a potent tool able to offer reliable results with lower efforts and costs than Sanger or individual-barcoded NGS ( Figure 1 ). The validness of the technique was also confirmed by the detection of mutations previously found by Sanger sequencing in the control patients. NGS and posterior case-mutation association revealed novel interesting results in the previously undiagnosed patients. A total of five variants in the SLC4A1 gene were detected by NGS in patients 122, 306, 323, and 493 ( Table 2 ). Patients 122 and 323 were carriers of a de novo mutation in the SLC4A1 p.589 hotspot position (R589C and R5889H, respectively), and patient 493 harbor the G609R mutation in the same gene, segregating with DRTA. It is interesting to note that these three patients carrying SLC4A1 mutations, 122, 323, and 493, were clinically diagnosed of DRTA at 13, 10, and 8 y of life, respectively, much older than the rest of the patients. Thus, our study describes, for the first time, DRTA cases in Spanish population caused by SLC4A1 gene mutations and supports the previously proposed phenotype–genotype correlation (18,19,20), suggesting that cases with late clinical onset DRTA are mutant for the SLC4A1 gene. It has been classically believed that loss-of-function mutations in the ATP6V1B1 gene caused DRTA with SNHL, whereas mutations in the ATP6V0A4 were responsible for cases without SNHL (2). This concept was challenged by Vargas-Poussou et al. (16) who in 2006 demonstrated genetic heterogeneity in DRTA associated with SNHL. These authors performed linkage studies to identify the disease locus in consanguineous families and then used the age of onset of SNHL to determine which gene to test first in the nonconsanguineous families. In 2013, Elhayek et al. (21) posed again the question whether ATP6V0A4 mutations could be responsible for early deafness, and reversely, ATP6V1B1 mutations could induce DRTA without hearing loss at the moment of diagnosis. In our study, we found three patients with mutations in ATP6V0A4 or ATP6V1B1 genes ( Table 2 ). Patient 306 had a heterozygous R394Q (16) mutation in the ATP6V1B1 gene. She was not deaf at diagnosis, and her hearing remained normal during the follow-up, up to date. Thus, these findings support that the association with SNHL is not a good indicator of the underlying causal gene, corroborating previously reported data (22,23,24), arising the advantage of NGS approach instead of selected Sanger phenotype–genotype sequencing approach ( Figure 1 ). It is interesting to note that patient 306 that presented early DRTA and was diagnosed at 2 mo of life was found to harbor only one variation in heterozygosis -R394Q- in the ATP6V1B1 gene. Segregation study revealed that none of her parents had the mutation. To our knowledge, this case represents the first de novo mutation in the ATP6V1B1 gene so far reported in the literature. The rest of the gene was analyzed by Sanger sequencing, and no variation susceptible of being assumed as mutation was found. However, NGS revealed a putative mutation, E508K, in heterozygosis at the SLC4A1 gene, which has been recognized as deleterious by SIFT and PolyPhen software, likely as a result of its location in a zone of overlapping protein domains. Interestingly, the patient’s mother harbored this mutation in heterozygosis and never presented DRTA symptoms so that the combined effect of the two genes’ mutations in heterozygosis seem to be required to cause clinical manifestations. It is of note that this variation has not been included as responsible for DRTA or spherocytosis diseases, but it was assumed as a somatic mutation in human cancers by the COSMIC project. There is the possibility of a potential undetected deletion involving one or more exons of the ATP6V1B1 gene. However, there is no evidence of the existence of large rearrangements in this gene (25). It is also worth noting that few Mendelian recessive diseases that are multigenic usually present mutations in heterozygosis located at different genes (26). So, we suggest that DRTA of patient 306 was probably caused by a combined effect of two mutations in two different genes: the R394Q mutation in the ATP6V1B1 gene and the E508K mutation in the SLC4A1 gene. Patient 158 had the p.P396fs mutation in the ATP6V0A4 gene and not SNHL. At diagnosis, this boy did not have hearing loss. Case 786 was carrying two splicing mutations at ATP6V0A4 gene: Intron 16+2 insA (2) and the Intron 7−2 T>C (pathogenicity corroborated by the Alternative Splice Site Predictor). This boy presented early SHNL diagnosed at few weeks of life.

Comparison among Sanger sequencing, NGS single-barcoded DNA, and next-generation sequencing (NGS) pool-barcoded DNA of the 10 patients involved in this study, taking as relative costs the pooling methodology. The pathway marked by the black arrows was followed in this study.
In case 491, no mutations were found by Sanger sequencing or NGS in any of the three analyzed genes. The clinical diagnosis of DRTA in this patient is supported by the presence of hypercloremic metabolic acidosis during a long-term follow-up when alkali treatment was transiently withdrawn and by the presence of SNHL and nephrocalcinosis. It is interesting to note that patient 491 was not hypokalemic, unlike the rest of the patients. Hypokalemia is a characteristic associated finding of patients with primary DRTA. Its underlying pathophysiologic mechanism remains to be fully understood, but it has been suggested that the degree of hypokalemia might depend on whether the disease is autosomal recessive or dominant. The most severe hypokalemia usually has been documented in patients with mutations in the genes encoding the ATP6B1 and ATP6V0A4 subunits of the H+ ATPase pump (27). Unfortunately, our series is too small to provide useful information on the relationship between the severity of hypokalemia and the underlying gene defect responsible for DRTA. Although the possibility of mutations occurring in a non-analyzed regulatory element like promoters (28) or introns (16) or the existence of large rearrangements (24) undetected by these techniques cannot be excluded, the association of another gene involved in the renal regulation of acid–base equilibrium as responsible for DRTA cannot be ruled out. Sanger sequencing of ATP6V1B1, ATP6V0A4, and SLC4A1 does not disclose mutations in up to 20% of patients with clinical diagnosis of primary DRTA which supports the existence of other candidate genes as potentially responsible for a significant number of primary DRTA cases. Whole exon sequencing may be necessary to find out the molecular cause of DRTA in this patient.
This study describes NGS as a useful diagnostic strategy to facilitate the molecular diagnosis of patients with primary DRTA, raises important considerations on the underlying genes and the disease’s phenotype, reports for the first time SLC4A1 gene mutations in Spanish individuals, suggests the possibility of combined heterozygous DRTA due to mutations in two different genes, and proposes those genetically unsolved cases as a target for the finding of new genes responsible for primary DRTA.
Methods
The study was based on 10 patients diagnosed with DRTA ( Table 1 ), recruited through the RenalTube consortium (12). All the patients or their parents signed an informed consent which was approved by the Regional Ethical Committee of Clinical Research from Hospital Universitario Central de Asturias, to participate in the study. Patients 311, 414, and 600 were Sanger sequenced for the ATP6V0A4 and ATP6V1B1 coding exons, and the identified mutations were used to validate the NGS procedure. The DNA from each patient was adjusted to 10 ng/µl using Real Time Taqman quantification with RNase P Detection Reagents of Life Technologies (Carlsbad, CA). Two pools of five patients were created A two-tube multiplex amplification for the whole-coding sequence plus at least 10 intronic flanking nucleotides (~8 kb) of ATP6V1B1, ATP6V0A4, and SLC4A1 were designed online with the Ion AmpliSeq Designer software (Life Technologies). Primer pairs to amplify a total of 67 fragments were provided by the manufacturer in two tubes (Life Technologies). The amplicons covered 99% of the target sequence (Supplementary Table S1 online). Each DNA pool was amplified with the Ion Ampliseq Library Kit 2.0 in conjunction with Ion Ampliseq Custom Primer Pool protocols, followed by template preparation by Ion PGM Template OT2 200 kit, and sequenced in a PGM sequencer using Ion PGM Sequencing 200 kit v2 according to the manufacturer procedures (Life Technologies) (29). Template-positive spheres were recovered using Dynabeads MyOne Streptavidin C1 beads and quantified using the Ion Sphere quality control assay with the Qubit 2.0 fluorometer (Life Technologies). Sphere particles were loaded in a 318v2 semiconductor chip and sequenced using a 500-flow runs, which support a template read-length of ~200 bp. The raw PGM data were processed using the Ion Torrent platform-specific pipeline software Torrent Suite v4.2 (Life Technologies) to generate sequence reads filtered according to the pipeline software quality controls and to remove poor signal reads. Reads assembling and variant identification were performed with the Variant Caller v4.2 software using FastQ files containing sequence reads and the Ion Ampliseq Designer BED file software to map the amplicons. Integrative Genome Viewer (Broad Institute, Cambridge, MA) was used for the analysis of depth coverage, sequences quality, and variants identification. The Variant Caller algorithm was set at threshold frequency of 5% to identify the nucleotide variants. For each putative mutation identified in one pool, each DNA used to create the pool was amplified for the corresponding exon and sequenced with BigDye chemistry (Life Technologies) using ABI3130xl equipment (Life Technologies) to identify the mutation carrier. The probable pathogenic outcome of novel missense and splicing mutations was analyzed in silico using SIFT and Polyphen-2 software pages based on sequence homology and physical properties of substituted amino acids, and Alternative Splice Site Predictor for the evaluation of consequences over possible splice donor or acceptor sites.
Statement of Financial Support
This research study was supported by PI09/90888, PI11/01672, and PI14/00702 grants integrated into the plan estalal of I+D+I 2013–2016 and funded by ISCIII and Fondo Europeo de Desarrollo Regional, GRUPIN 14-020 grant from the Hospital Universitario Central de Asturias, and the Fundación Bancaria CajAstur (Spain).
Disclosure
There are no conflicts of interest.
References
OMIM. Online Mendelian Inheritance in Man, 1993. (http://www.omim.org.)
Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 1999;21:84–90.
Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 2002;39:796–803.
Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant 2012;27:3691–704.
Tanphaichitr VS, Sumboonnanonda A, Ideguchi H, et al. Novel AE1 mutations in recessive distal renal tubular acidosis. Loss-of-function is rescued by glycophorin A. J Clin Invest 1998;102:2173–9.
Bruce LJ, Unwin RJ, Wrong O, Tanner MJ. The association between familial distal renal tubular acidosis and mutations in the red cell anion exchanger (band 3, AE1) gene. Biochem Cell Biol 1998;76:723–8.
Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr 2014;164:691–698.e1.
Chu CY, King J, Berrini M, et al. Degradation mechanism of a Golgi-retained distal renal tubular acidosis mutant of the kidney anion exchanger 1 in renal cells. Am J Physiol Cell Physiol 2014;307:C296–307.
Renkema KY, Stokman MF, Giles RH, Knoers NV. Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol 2014;10:433–44.
Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011;475:348–52.
Tavira B, Gómez J, Málaga S, et al. A labor and cost effective next generation sequencing of PKHD1 in autosomal recessive polycystic kidney disease patients. Gene 2015;561:165–9.
Renaltube. Primary Tubular Disorders Database, 2014. (http://www.renaltube.com.)
Sritippayawan S, Kirdpon S, Vasuvattakul S, et al. A de novo R589C mutation of anion exchanger 1 causing distal renal tubular acidosis. Pediatr Nephrol 2003;18:644–8.
Yenchitsomanus PT. Human anion exchanger1 mutations and distal renal tubular acidosis. Southeast Asian J Trop Med Public Health 2003;34:651–8.
Rungroj N, Devonald MA, Cuthbert AW, et al. A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells. J Biol Chem 2004;279:13833–8.
Vargas-Poussou R, Houillier P, Le Pottier N, et al. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 2006;17:1437–43.
Loman NJ, Misra RV, Dallman TJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 2012;30:434–9.
Karet FE, Gainza FJ, Györy AZ, et al. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA 1998;95:6337–42.
Jarolim P, Shayakul C, Prabakaran D, et al. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band 3) Cl-/HCO3- exchanger. J Biol Chem 1998;273:6380–8.
Weber S, Soergel M, Jeck N, Konrad M. Atypical distal renal tubular acidosis confirmed by mutation analysis. Pediatr Nephrol 2000;15:201–4.
Elhayek D, Perez de Nanclares G, Chouchane S, et al. Molecular diagnosis of distal renal tubular acidosis in Tunisian patients: proposed algorithm for Northern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes. BMC Med Genet 2013;14:119.
Andreucci E, Bianchi B, Carboni I, et al. Inner ear abnormalities in four patients with dRTA and SNHL: clinical and genetic heterogeneity. Pediatr Nephrol 2009;24:2147–53.
Kose E, Sirin Kose S, Alparslan C, et al. Val2Ala mutation in the Atp6v0a4 gene causes early-onset sensorineural hearing loss in children with recessive distal renal tubular acidosis: a case report. Ren Fail 2014;36:808–10.
Gao Y, Xu Y, Li Q, Lang Y, Dong Q, Shao L. Mutation analysis and audiologic assessment in six Chinese children with primary distal renal tubular acidosis. Ren Fail 2014;36:1226–32.
Miura K, Sekine T, Takahashi K, et al. Mutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol Dial Transplant 2013;28:2123–30.
Chang L, Yuan W, Zeng H, et al. Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients. BMC Med Genomics 2014;7:24.
Batlle D, Moorthi KM, Schlueter W, et al. Distal renal tubular acidosis and the potassium enigma. Semin Nephrol 2006;26:471–8.
Myers RM, Tilly K, Maniatis T. Fine structure genetic analysis of a beta-globin promoter. Science 1986;232:613–8.
Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun 2014;5:5326.
Acknowledgements
The authors thank the Renaltube group, because of its labor facilitating the recruitment of patients with DRTA. The authors also thank the Scientific-Technical Services of the University of Oviedo because of the collaboration in processing NGS data.
Author information
Authors and Affiliations
Consortia
Corresponding author
Supplementary information
Supplementary Figures
(PDF 546 kb)
Supplementary Tables
(DOC 33 kb)
PowerPoint slides
Rights and permissions
About this article

Cite this article
Gómez, J., Gil-Peña, H., Santos, F. et al. Primary distal renal tubular acidosis: novel findings in patients studied by next-generation sequencing. Pediatr Res 79, 496–501 (2016). https://doi.org/10.1038/pr.2015.243
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.243
This article is cited by
-
Distal renal tubular acidosis: a systematic approach from diagnosis to treatment
Journal of Nephrology (2021)
-
Distal renal tubular acidosis. Clinical manifestations in patients with different underlying gene mutations
Pediatric Nephrology (2018)
-
An in vitro splicing assay reveals the pathogenicity of a novel intronic variant in ATP6V0A4 for autosomal recessive distal renal tubular acidosis
BMC Nephrology (2017)
