
Abstract
We review recent findings and hypotheses on the roles of gut microbiome in the pathogenesis of nonalcoholic fatty liver diseases (NAFLD). Microbial metabolites and cell components contribute to the development of hepatic steatosis and inflammation, key components of nonalcoholic steatohepatitis (NASH), the severe form of NAFLD. Altered gut microbiome can independently cause obesity, the most important risk factor for NAFLD. This capability is attributed to short-chain fatty acids (SCFAs), major gut microbial fermentation products. SCFAs account for a large portion of caloric intake of the host, and they enhance intestinal absorption by activating GLP-2 signaling. However, elevated SCFAs may be an adaptive measure to suppress colitis, which could be a higher priority than imbalanced calorie intake. The microbiome of NASH patients features an elevated capacity for alcohol production. The pathomechanisms for alcoholic steatohepatitis may apply to NASH. NAFLD/NASH is associated with elevated Gram-negative microbiome and endotoxemia. However, many NASH patients exhibited normal serum endotoxin indicating that endotoxemia is not required for the pathogenesis of NASH. These observations suggest that microbial intervention may benefit NAFLD/NASH patients. However, very limited effects were observed using traditional probiotic species. Novel probiotic therapy based on NAFLD/NASH specific microbial composition represents a promising future direction.
Similar content being viewed by others

Main
Human microbiome, especially the gut microbiome, has gained attention recently due to the advancement of the high-throughput “next generation sequencing techniques.” Previous studies employing culture dependent techniques were labor intensive and inevitably biased as 60–80% of the gut microbes cannot be cultivated (1). With the high throughput culture-independent sequencing techniques, taxonomic assignments of the uncultured, undefined microbes into operational taxonomic units (OTUs) are based on clustering of the 16S rRNA sequences (18S rRNA microbiome are not discussed in this review as eukaryotes are rare in the gut microbiome and there are few published studies.). Frequently, microbial OTUs are grouped at 97% similarity in 16S rRNA sequences and such OTUs are considered equivalent to species. On average, thousands of OTUs are observed in a human gut microbiota (2), which is substantially higher than the previous estimations of 300–500 species based on culture-dependent studies (3).
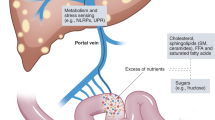
Normal gut microbes benefit human health through various mechanisms such as the digestion of fibers and resistant starch, synthesis of vitamins, prevention of pathogen colonization, and immunomodulation in the development of intestinal mucosal immunity (4). This list will likely grow when the uncultured, undefined species are characterized. On the other hand, many human diseases are associated with altered gut microbiome and in some cases causal relationships were observed, such as Clostridium difficile and diarrhea (5). In many circumstances, the disrupted gut microbiome could have an adverse effect on liver health as the bacterial metabolites and the bacterial cell components are drained to the liver through portal vein. Here, we review recent progress in understanding the associations between altered gut microbiota and fatty liver diseases with particular attention to adolescent patients. Evidence shows that microbial products contribute to the development or maintenance of liver steatosis and inflammation, two key components for the diagnosis of nonalcoholic steatohepatitis (NASH), the severe form of nonalcoholic fatty liver diseases (NAFLD) now a major cause for liver transplantation (6).
Short-Chain Fatty Acids and NAFLD
Hypothetical Mechanism 1: Elevated Energy Extraction in the Form of Short-Chain Fatty Acids
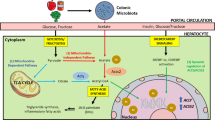
Short-chain fatty acids (SCFAs) are the major fermentation products of gut microbes. In the human colon, the normal gut microbiome produces a concentration of 50–100 mmol/l SCFAs (7), mainly comprised of acetate, propionate and butyrate. These SCFAs provide a sizable portion of the energy needs of the human body. Research using germ-free animals indicated that the SCFAs produced by the microbes account for ~30% of energy extraction from the diet (8).
SCFAs are implicated in the pathogenesis of NAFLD because of their possible contribution to obesity. Initial evidence for an altered gut microbiota in obesity is the observation by Ley et al. that ob/ob mice (9) and obese patients (10) exhibit reduced abundance of Bacteroidetes and proportionally increased abundance of Firmicutes. Altered gut microbiota in obesity was also observed by other researchers, although the observed changes were usually different (11,12,13,14,15,16,17,18), and frequently opposite (12,13,14,15,16,17,18) to the observations of Ley et al. Note that only three studies stated that the enrolled patients and controls did not take antibiotics in the 3-mo period before sample collection; and all three studies reported an increased Bacteroidetes/Firmicutes ratio in the gut of obese patients (12,13,14,19) ( Table 1 ). Our group paid particular attention to proton pump inhibitors and histamine receptor antagonists (14,19) as these agents are known to affect Firmicutes (20). These precautions for patient enrollment gave us confidence in the observed changes in the microbiome of our pediatric NASH patients.
Currently, the prevalent hypothetical mechanism for the role of altered gut microbiota in the development of obesity is that the gut microbiota of obese animals have increased capacity to harvest energy. The first evidence was provided by Turnbaugh et al. (21): (i) the gut microbiome of the ob/ob mice is enriched in carbohydrate metabolizing genes; (ii) the cecum of these animals has an increased concentration of SCFAs; (iii) the stool of these animals retains significantly less energy than their lean mates according to bomb calorimetry; and (iv) most strikingly, transplantation of the germ free mice with the gut microbiome from ob/ob mice caused greater fat gain than transplants from lean animals. In humans, increased production of SCFAs by the gut microbiota was also observed in overweight and obese people, compared to lean subjects (13). The conflict here is that while Turnbaugh et al. observed decreased Bacteroidetes/Firmicutes ratio in the gut of obese mice and obese humans, Schwiertz et al. reported increased Bacteroidetes/Firmicutes ratio in the gut of obese people. Although members in both phyla produce SCFAs, it seems that Bacteroidetes (e.g., Prevotella) produce more SCFAs (22). Prevotella is well equipped for the production of SCFAs from polysaccharide metabolism. For example, the P. bryantii genome encodes 203 carbohydrate active enzymes including 107 glycoside hydrolases, 53 glycosyl transferases, 14 polysaccharide lyases, and 19 carbohydrate esterases (23).
Our recent effort in deciphering the gut microbial composition of obese children revealed that Bacteroidetes were significantly elevated (mainly Prevotella) in obese and NASH patients, compared to lean healthy children (14). Although our cross-sectional study supports a causal relationship between high Prevotella and obesity, this hypothetical mechanism needs to be tested in a longitudinal study. One possible strategy is to use gnotobiotic mice carrying the gut microbiome of pediatric obese patients or of lean children, and examine their differential influence on host metabolism and weight gain.
What caused the increased representation of Prevotella? According to De Filippo et al. (22), a diet high in plant fibers provides more fermentation substrates for Prevotella and therefore stimulates its proliferation. On the other hand, high fat diets promote the growth of Firmicutes in mice (24). A comprehensive study investigating the correlation of dietary components with human gut microbial composition reported similar results (25).
Hypothetical Mechanism 2: Intestinotrophic Effect of SCFAs Mediated by Glucagon-Like Peptide 2
Initial evidence for SCFAs enhancing nutritional absorption came from studies on total parenteral nutrition (TPN) for short-bowel syndrome (SBS). Supplementation of SCFAs to TPN prevented TPN associated mucosal atrophy (26) and increased the intestinal absorption of glucose (27). The underlying mechanisms include the induction of nutrient transporters such as GLUT2 in enterocytes (27,28), the induction of enterocyte proliferation and the inhibition of apoptosis (29). Tappenden et al. also noticed that SCFA supplementation induced the expression of ileal proglucagon mRNA and plasma glucagon-like peptide 2 (GLP-2), establishing the current hypothesis that the intestinotrophic effects of SCFAs are mediated by GLP-2 (28,30). Since SCFAs are elevated in the gut of obese patients, they may contribute to the development/maintenance of obesity through elevated intestinal absorption.
However, the idea of targeting SCFA/GLP-2 mediated intestinal absorption in the treatment of obesity and related complications is undermined by the report that increased GLP-2 levels, achieved by microbial intervention or subcutaneous administration of GLP-2 (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33) is beneficial for ob/ob mice (31). The beneficial effect is the improvement of the leaky gut (reduced permeability), which leads to lower plasma lipopolysaccharide (LPS) and cytokines levels, and consequently a decreased hepatic expression of oxidative stress and inflammatory markers. The knowledge gap here is that while the study of Cani et al. is suggestive of a lower level of GLP-2 in ob/ob mice, this is not consistent with the reports that SCFAs are high in the gut of ob/ob mice (21) and SCFAs induce GLP-2 (28,30). Further studies on the interaction between SCFA and GLP-2 and the underlying mechanism may provide an explanation for this inconsistency.
Hypothetical Mechanism 3: Suppression of Colitis Through GPR43 Mediated Signaling
Because colitis enhances gut permeability, and consequently allows elevated hepatic exposure of microbial products via portal circulation, suppression of colitis by SCFAs could exert a major protection for liver health. Human inflammatory bowel diseases are associated with reduced production of SCFAs in the gut (32). Also, beneficial effects of SCFA supplementation on colitis have been observed in many different studies (e.g., ref. 33). Although the effectiveness of the SCFA therapy may be explained by the intestinotrophic effect of SCFAs mediated by GLP-2, evidence also suggests that the G-protein coupled receptor 43 (GPR43), a receptor for SCFAs, suppresses inflammatory responses in the gut. Maslowski et al. examined the effect of SCFAs/GPR43 on intestinal inflammatory responses using a dextran sulphate sodium (DSS) induced colitis model (34). The DSS induced colitis is more severe in germ-free mice than in conventionally raised animals, and the colitis was ameliorated by acetate supplementation. The beneficial effect of acetate is absent in GPR43−/− mice. By suppressing colitis, SCFAs/GPR43 signaling helps to improve gut permeability and therefore minimize the hepatic injuries imposed by microbial cell components and microbial products. The activity of SCFAs/GRP43 is not limited to the intestines. Kimura et al. showed in mice that SCFAs/GPR43 suppresses insulin signaling in adipose tissue, leading to inhibited fat accumulation in adipose tissue (35). These authors demonstrated that GPR43 deficient mice are obese on a normal diet, whereas mice overexpressing GPR43 specifically in adipose tissue remain lean when fed a high-fat diet.
A Summary on SCFAs and Liver Disease
Current literature suggests that SCFAs have opposing effects on liver health: on one side, SCFAs account for ~30% of calorie intake, and, the intestinotrophic effect of SCFAs mediated by GLP-2 enhances the intestinal absorption, both mechanisms may contribute to the development of obesity and obesity related phenotypes including liver diseases. On the other side, through their receptor GPR43, SCFAs suppress colon inflammation, therefore protect liver from the insults of microbial cell components coming from the portal vein; SCFAs/GPR43 may also benefit liver health by down-regulating insulin signaling in adipose tissue. The seemingly conflicting roles of SCFAs on liver health could be a consequence of imbalanced calorie intake/expenditure associated with modern life styles. When balance is achieved between caloric intake and expenditure, SCFAs would only benefit liver health.
Alcohol and NAFLD
Gut Microbes Produce Alcohol
It is well known that the normal gut microbiome produces alcohol, as suggested initially by the increased blood alcohol level after intake of alcohol free food (36). Production of ethanol in the gut is also reflected by the fact that human liver and gastrointestinal tract have the highest activities of alcohol dehydrogenases in the body (37). Because these enzymes are constantly metabolizing alcohol, it is difficult to assess the quantity of alcohol produced in the gut. Nevertheless, in vitro studies demonstrated the alcohol producing capacity of some gut microbial species. For example, under anaerobic conditions, 1 g (wet weight) of E. coli produces 0.8 g ethanol in 1 h (38).
The Rise, Fall, and Rise Again of the “Alcohol Hypothesis”: Our Gene Expression Study
The possible role for endogenous alcohol in NAFLD was first implicated in ob/ob mice. Cope et al. found that the alcohol in the breath of obese animals is higher than that of lean animals (39). This observation prompted the authors to propose the “alcohol hypothesis” of NASH. However, the same researchers could not find any difference in the breath alcohol concentration between NASH patients and lean controls (40). In 2009, our group completed the first global gene expression analysis of NASH livers and normal controls (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17470), and found that many of the alcohol and aldehyde dehydrogenases are among the most up-regulated genes in NASH livers (41). Additionally, other alcohol metabolizing mechanisms including CYP2E1 and catalase are also highly elevated in NASH livers. We confirmed these observations with quantitative real-time PCR (qRT-PCR) and western blots and are convinced that all liver pathways for metabolizing alcohol were highly elevated in NASH livers. The most reasonable explanation for these observations is that alcohol was elevated in the circulation of the NASH patients and caused up-regulation of the alcohol metabolizing capacity.
More About Alcohol Related Gene Expression in NAFLD: CYP2E1
Induced CYP2E1 in NASH liver was reported as early as in 1998 (42) and confirmed in another study (43). Similar observations were reported in a methionine and choline deficient diet (MCDD) animal model of NASH (44,45). CYP2E1, a major component of the microsomal ethanol oxidizing system (MEOS), catalyzes ethanol oxidation and produces free radicals that may lead to liver inflammation (46). CYP2E1 is an ethanol inducible enzyme (47,48), and is well known for its role in the pathogenesis of alcoholic steatohepatitis. Therefore, the observation of elevated CYP2E1 in NASH liver led to the hypothesis that free radicals produced by CYP2E1 may play a critical role in the pathogenesis of NASH (42,43). However, these investigators did not discuss the possible cause of elevated CYP2E1 activity. One explanation for induced CYP2E1 in NASH livers is that CYP2E1 may be induced by fatty acid as demonstrated in in vitro studies (49). However, in intact animals, high fat diets did not induce CYP2E1, but induced other components of MEOS (50). Therefore, in our opinion, induced CYP2E1 in NASH livers is more likely a consequence of elevated alcohol in the circulation.
Alcohol, Gut Microbiome, and NASH
Although Nair et al. did not observe an elevated breath alcohol in NAFLD patients (40), the “alcohol hypothesis” continued. Volynets et al. observed elevated serum alcohol concentration in adult NAFLD patients (51). While occasional drinking might influence the assessment of serum alcohol with adult NAFLD patients, it is less a concern with pediatric patients. We reported that pediatric NASH patients exhibited higher serum alcohol concentrations than those of healthy controls and non-NASH obese patients (14). Since we have demonstrated elevated alcohol scavenging capabilities in NASH livers, it is conceivable that the difference in peripheral serum alcohol between NASH patients and normals is much diminished, compared to that in portal blood.
After a thorough analysis of the dietary intake, we excluded the possible contributions of alcoholic drinks, fruit or aspartame on elevated serum alcohol, and arrived at the hypothesis that the gut microbiome is responsible for elevated endogenous alcohol in NASH patients. The gut microbiomes from NASH patients, obese patients and healthy controls were quantitatively assessed by 16S rRNA pyrosequencing (14). Abundant differences at all taxonomic levels and ecological diversities were found between NASH patients and healthy controls, and between obese patients and healthy controls. The microbiome composition of NASH patients and obese patients are quite similar except that they differ significantly in the abundance of Escherichia, a genus of the Enterobacteriaceae family. Enterobacteriaceae exhibits a mixed-acid fermentation pathway, a major product of which is ethanol (38,52,53,54). These studies demonstrated the alcohol producing capabilities of Enterobacteriaceae members in vitro (38,52,53) and in vivo (54).
Summary and Future Directions for the Alcohol Hypothesis
Available data support a role for endogenous alcohol in the pathogenesis of NASH. Alcohol produced by the gut microbes may contribute to the development of NASH by mechanisms similar to those described for alcoholic steatohepatitis. Currently, all studies in support of the “alcohol hypothesis” were cross-sectional and do not support a causal relationship. Prospective, time-course studies are needed to prove a causal relationship between altered gut microbiome and increased alcohol production. Additionally, it is of particular interest and importance to investigate the driving forces for the alterations in gut microbiome in NAFLD and NASH patients. Possible factors that lead to elevated abundance of alcohol producing Escherichia include high fat diet (55) and intake of sub-therapeutic levels of antibiotics (56).
LPS And NAFLD
Possible Roles of LPS in Metabolic Diseases and NAFLD/NASH: Animal Models
Many microbial cell components, or pathogen-associated molecular patterns (PAMPs) including lipopolysaccharide (LPS, endotoxin), flagellin, lipoteichoid acid, peptidoglycan, and unmethylated CpG motifs may affect the physiology and pathology of their host, mediated by Toll-like receptors or other pattern recognition receptors. Among these molecules, LPS is the most studied for possible role in liver diseases.
The first evidence in support of a role for LPS in the pathogenesis of NASH was the observation that endotoxemia readily induces steatohepatitis in obese rats and mice (57). With the methionine and choline deficient diet (MCDD) mouse model for NASH, Rivera et al. observed portal endotoxemia and elevated expression of Toll-like receptor 4, CD14 and MD2 in Kupffer cells (58). These genes are key components of the LPS/TLR4 signaling pathway leading to the production of pro-inflammatory cytokines (59). One mechanism for hepatic exposure to endotoxin is increased gut permeability such as colitis or celiac disease. Another is that high-fat diets may facilitate LPS uptake through elevated chylomicron production in intestinal epithelial cells (60).
More strikingly, Cani et al. introduced a novel concept that chronic metabolic endotoxemia (LPS concentration increased two to three times compared to normal) leads to metabolic syndrome (61). The authors introduced metabolic endotoxemia in mice through continuous subcutaneous infusion of LPS, which caused body weight gain, liver weight gain, adipose tissue weight gain, liver insulin resistance, fasting glycemia, and insulinemia, features that are similar to those induced by high-fat diets. These responses did not occur with CD14 mutant mice, indicating that CD14/TLR4 signaling is required for LPS induced metabolic diseases.
The importance of LPS/TLR4 signaling in the pathogenesis of NAFLD is strengthened by another study using a different animal model. Henao-Mejia et al. found that NLRP6 inflammasome deficiency leads to an altered transmissible, colitogenic gut microbiota (62). When fed with MCDD, these inflammasome deficient mice developed NASH with significantly higher severity than wild-type animals (63). They demonstrated that the colitogenic microbiota is responsible for the enhanced disease severity with a co-housing experiment. Furthermore, they demonstrated that TLR4 and TLR9 (but not TLR5) signaling are both required for enhanced NASH severity.
These animal studies suggest that endotoxin is sufficient to induce all NASH pathologies. How well do these animal studies reflect the pathology in NASH patients?
Possible Roles of LPS on Metabolic Diseases and NAFLD/NASH: Human Studies
Several groups reported elevated serum endotoxin in NAFLD and NASH patients (51,64,65,66), including pediatric patients (66). In line with these observations, NAFLD/NASH patients also exhibit elevated serum LPS binding protein (67) and LPS antibodies (68).
We examined possible correlations between NASH and the Gram-negative microbiomes, the source of LPS (19). We found that our adolescent NASH patients carry a gut microbiota with elevated abundance in Gram-negative bacteria, compared to healthy controls. Additionally, we also observed an elevated serum endotoxin level and increased incidence of endotoxemia in NASH patients. However, a closer examination of the data revealed no correlation between Gram-negative bacteria abundance and the concentration of serum endotoxin. Our data revealed that endotoxemia is absent from the majority of NASH patients; therefore, we conclude that endotoxemia is not required for the pathogenesis of NAFLD/NASH.
Our observation prompted us to revisit previous studies on endotoxemia in NAFLD/NASH. In the first study demonstrating the association between NAFLD and endotoxemia, 2 out of 12 NAFLD patients are free from endotoxemia (64). In the following years, several reports from the same group and others also demonstrated the association of NAFLD with endotoxemia (51,65,66). A common observation from these reports was the broad range of serum endotoxin levels in NAFLD patients. Normal serum endotoxin was observed in some of the NAFLD/NASH patients from different countries in different studies. Further evidence in support of normal endotoxin in some of the NAFLD/NASH patients include the observations that serum LPS antibody levels (68) and plasma LPS binding protein levels (67) of NASH patients are of broad ranges, overlapping with that of the normals. Overall, these studies suggest that, endotoxemia is not required in the pathogenesis of NASH. Endotoxemia may contribute to disease progression in some NASH cases, but other challenges such as gut derived alcohol (39,41), endoplasmic reticulum stress, and adipose tissue derived threats may cause NASH independently from endotoxemia. Our observations highlight the current “multi-hit” hypothesis for the pathogenesis of NASH (69).
Microbiome Intervention in the Treatment of NAFLD/NASH
The observations that the gut microbiota play critical roles in the pathogenesis of NAFLD/NASH lead to and support the hypothesis that microbiota manipulation is a potentially effective therapeutic option for NAFLD/NASH. Available interventions for altering gut microbiota include diet, antibiotics, probiotics, prebiotics, synbiotics, and microbiota transplantation. Among these, probiotics have been tested for the treatment of NAFLD/NASH in animal models and human subjects.
With several rodent NAFLD models, a probiotic mixture VSL#3 (containing Streptococcus, Bifidobacterium, and Lactobacillus) improved liver histology, reduced liver fat, and decreased serum ALT levels (70,71,72,73). With the MCDD mouse model, VSL#3 reduced liver fibrosis but aggravated liver fat and serum ALT (74).
Several human studies evaluated the therapeutic effect of probiotics on NAFLD/NASH. VSL#3 seems to reduce oxidative stress in NAFLD patients (75), but no control was included in this study. A randomized, double-blinded, placebo-controlled study was conducted to evaluate the effect of synbiotics including Bifidobacterium longum and fructo-oligosaccharides in NAFLD patients and reported small beneficial effects (76). In another randomized controlled trial examining the effects of probiotic therapy on NASH patients using Lactobacillus and Bifidobacterium spp. no significant difference in BMI, fasting glucose, cholesterol, or triglyceride was observed between the probiotics treated group and the control (18).
One major pitfall of these probiotics/synbiotics trials is that the probiotic species did not target NAFLD/NASH specific microbiota. Both Bifidobacterium and Lactobacillus are minor genera in the gut compared to Bacteroides, Prevotella, Blautia, or Faecalibacteria (14). Moreover, Lactobacillus tends to be increased in NASH patients (L. Zhu, R.D. Baker, and S.S. Baker, unpublished data). Since there is no deficiency of these traditional probiotic species in the gut of NAFLD/NASH patients, it is no surprise that supplementation with these species did not significantly alleviate microbiota related pathologies. Major taxa depleted in the gut of NASH patients are Lachnospiraceae species, Ruminococcaceae species, and Veillonellaceae species (14), from which effective probiotic species for NAFLD/NASH may be selected and evaluated in future studies.
Conclusions
Abundant evidence from NAFLD/NASH animal models and patients including pediatric patients revealed several possible mechanisms for gut microbiota to contribute to the development and maintenance of liver steatosis and hepatic inflammation. The gut microbial composition of the NAFLD/NASH patients deviates from that of healthy people. The altered microbiome in NAFLD/NASH may produce more SCFAs and alcohol, and carry more Gram-negative species that supply LPS to the portal circulation. Although elevated SCFAs may contribute to increased calorie intake, it may also be an adaptive action of the host to control inflammatory signaling. Therefore, gut microbial taxa associated with higher capacities in alcohol and LPS production may represent better targets for therapeutic interventions on the gut microbiome. Currently, most of the microbiome interventions on NAFLD/NASH patients used traditional probiotic species that are not depleted in the gut of the patients and exhibited very limited effect in clinical trials. Novel, disease targeting probiotic therapies may be designed based on the altered microbial composition of the NAFLD/NASH patients.
Statement of Financial Support
Work done in the authors’ laboratory is mainly supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (U01 DK061728, to S.S.B.; Bethesda, MD), the Peter and Tommy Fund, Inc., Buffalo, NY (to S.S.B. and L.Z.), and a departmental start-up fund (to L.Z.).
References
Suau A, Bonnet R, Sutren M, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 1999;65:4799–807.
Arumugam M, Raes J, Pelletier E, et al.; MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011;473:174–80.
Simon GL, Gorbach SL . Intestinal flora in health and disease. Gastroenterology 1984;86:174–93.
Flint HJ, Scott KP, Louis P, Duncan SH . The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 2012;9:577–89.
Brandt LJ, Aroniadis OC, Mellow M, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol 2012;107:1079–87.
Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA . Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011;141:1249–53.
Duncan SH, Louis P, Thomson JM, Flint HJ . The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 2009;11:2112–22.
Wostmann BS, Larkin C, Moriarty A, Bruckner-Kardoss E . Dietary intake, energy metabolism, and excretory losses of adult male germfree Wistar rats. Lab Anim Sci 1983;33:46–50.
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI . Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 2005;102:11070–5.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI . Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022–3.
Duncan SH, Lobley GE, Holtrop G, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–4.
Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA 2009;106:2365–70.
Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18:190–5.
Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601–9.
Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010;5:e9085.
Collado MC, Isolauri E, Laitinen K, Salminen S . Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 2008;88:894–9.
Kalliomäki M, Collado MC, Salminen S, Isolauri E . Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr 2008;87:534–8.
Wong VW, Tse CH, Lam TT, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis–a longitudinal study. PLoS ONE 2013;8:e62885.
Yuan J, Baker SS, Liu W, et al. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J Gastroenterol Hepatol 2014;29:1292–8.
Howell MD, Novack V, Grgurich P, et al. Iatrogenic gastric acid suppression and the risk of nosocomial Clostridium difficile infection. Arch Intern Med 2010;170:784–90.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI . An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–31.
De Filippo C, Cavalieri D, Di Paola M, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 2010;107:14691–6.
Purushe J, Fouts DE, Morrison M, et al.; North American Consortium for Rumen Bacteria. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol 2010;60:721–9.
Murphy EF, Cotter PD, Healy S, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 2010;59:1635–42.
Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011;334:105–8.
Koruda MJ, Rolandelli RH, Bliss DZ, Hastings J, Rombeau JL, Settle RG . Parenteral nutrition supplemented with short-chain fatty acids: effect on the small-bowel mucosa in normal rats. Am J Clin Nutr 1990;51:685–9.
Tappenden KA, Thomson AB, Wild GE, McBurney MI . Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats. Gastroenterology 1997;112:792–802.
Tappenden KA, McBurney MI . Systemic short-chain fatty acids rapidly alter gastrointestinal structure, function, and expression of early response genes. Dig Dis Sci 1998;43:1526–36.
Bartholome AL, Albin DM, Baker DH, Holst JJ, Tappenden KA . Supplementation of total parenteral nutrition with butyrate acutely increases structural aspects of intestinal adaptation after an 80% jejunoileal resection in neonatal piglets. JPEN J Parenter Enteral Nutr 2004;28:210–22; discussion 222–3.
Tappenden KA, Drozdowski LA, Thomson AB, McBurney MI . Short-chain fatty acid-supplemented total parenteral nutrition alters intestinal structure, glucose transporter 2 (GLUT2) mRNA and protein, and proglucagon mRNA abundance in normal rats. Am J Clin Nutr 1998;68:118–25.
Cani PD, Possemiers S, Van de Wiele T, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009;58:1091–103.
Treem WR, Ahsan N, Shoup M, Hyams JS . Fecal short-chain fatty acids in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 1994;18:159–64.
Scheppach W . Treatment of distal ulcerative colitis with short-chain fatty acid enemas. A placebo-controlled trial. German-Austrian SCFA Study Group. Dig Dis Sci 1996;41:2254–9.
Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009;461:1282–6.
Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 2013;4:1829.
Blomstrand R . Observations of the formation of ethanol in the intestinal tract in man. Life Sci II 1971;10:575–82.
Engeland K, Maret W . Extrahepatic, differential expression of four classes of human alcohol dehydrogenase. Biochem Biophys Res Commun 1993;193:47–53.
Dawes EA, Foster SM . The formation of ethanol in Escherichia coli. Biochim Biophys Acta 1956;22:253–65.
Cope K, Risby T, Diehl AM . Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology 2000;119:1340–7.
Nair S, Cope K, Risby TH, Diehl AM, Terence RH . Obesity and female gender increase breath ethanol concentration: potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am J Gastroenterol 2001;96:1200–4.
Baker SS, Baker RD, Liu W, Nowak NJ, Zhu L . Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010;5:e9570.
Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C . Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998;27:128–33.
Chalasani N, Gorski JC, Asghar MS, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003;37:544–50.
Weltman MD, Farrell GC, Liddle C . Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996;111:1645–53.
Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR . CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest 2000;105:1067–75.
Lieber CS . Microsomal ethanol-oxidizing system (MEOS): the first 30 years (1968-1998)–a review. Alcohol Clin Exp Res 1999;23:991–1007.
Takahashi T, Lasker JM, Rosman AS, Lieber CS . Induction of cytochrome P-4502E1 in the human liver by ethanol is caused by a corresponding increase in encoding messenger RNA. Hepatology 1993;17:236–45.
Ingelman-Sundberg M, Johansson I, Yin H, et al. Ethanol-inducible cytochrome P4502E1: genetic polymorphism, regulation, and possible role in the etiology of alcohol-induced liver disease. Alcohol 1993;10:447–52.
Raucy JL, Lasker J, Ozaki K, Zoleta V . Regulation of CYP2E1 by ethanol and palmitic acid and CYP4A11 by clofibrate in primary cultures of human hepatocytes. Toxicol Sci 2004;79:233–41.
Lieber CS, Lasker JM, DeCarli LM, Saeli J, Wojtowicz T . Role of acetone, dietary fat and total energy intake in induction of hepatic microsomal ethanol oxidizing system. J Pharmacol Exp Ther 1988;247:791–5.
Volynets V, Küper MA, Strahl S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci 2012;57:1932–41.
Paege LM, Gibbs M . Anaerobic dissimilation of glucose-C14 by Escherichia coli. J Bacteriol 1961;81:107–10.
Clark DP . The fermentation pathways of Escherichia coli. FEMS Microbiol Rev 1989;5:223–34.
Brooks JB, Basta MT, el Kholy AM . Studies of metabolites in diarrheal stool specimens containing Shigella species by frequency-pulsed electron capture gas-liquid chromatography. J Clin Microbiol 1985;21:599–606.
Yin X, Peng J, Zhao L, et al. Structural changes of gut microbiota in a rat non-alcoholic fatty liver disease model treated with a Chinese herbal formula. Syst Appl Microbiol 2013;36:188–96.
Looft T, Johnson TA, Allen HK, et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci USA 2012;109:1691–6.
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM . Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 1997;94:2557–62.
Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M . Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 2007;47:571–9.
Testro AG, Visvanathan K . Toll-like receptors and their role in gastrointestinal disease. J Gastroenterol Hepatol 2009;24:943–54.
Laugerette F, Vors C, Géloën A, et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem 2011;22:53–9.
Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–72.
Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014;156:1045–59.
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179–85.
Thuy S, Ladurner R, Volynets V, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr 2008;138:1452–5.
Harte AL, da Silva NF, Creely SJ, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 2010;7:15.
Alisi A, Manco M, Devito R, Piemonte F, Nobili V . Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2010;50:645–9.
Ruiz AG, Casafont F, Crespo J, et al. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg 2007;17:1374–80.
Verdam FJ, Rensen SS, Driessen A, Greve JW, Buurman WA . Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J Clin Gastroenterol 2011;45:149–52.
Tilg H, Moschen AR . Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836–46.
Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37:343–50.
Ma X, Hua J, Li Z . Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol 2008;49:821–30.
Esposito E, Iacono A, Bianco G, et al. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J Nutr 2009;139:905–11.
Mencarelli A, Cipriani S, Renga B, et al. VSL#3 resets insulin signaling and protects against NASH and atherosclerosis in a model of genetic dyslipidemia and intestinal inflammation. PLoS ONE 2012;7:e45425.
Velayudham A, Dolganiuc A, Ellis M, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009;49:989–97.
Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol 2005;39:540–3.
Malaguarnera M, Vacante M, Antic T, et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci 2012;57:545–53.
Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480–4.
Armougom F, Henry M, Vialettes B, et al. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One 2009;4:e7125.
Santacruz A, Collado MC, Garcia-Valdes L, et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 2010;104:83–92.
Duncan SH, Belenguer A, Holtrop G, et al. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol 2007;73:1073–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhu, L., Baker, R. & Baker, S. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr Res 77, 245–251 (2015). https://doi.org/10.1038/pr.2014.157
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2014.157
This article is cited by
-
Gut microbiome features and metabolites in non-alcoholic fatty liver disease among community-dwelling middle-aged and older adults
BMC Medicine (2024)
-
Pivotal Dominant Bacteria Ratio and Metabolites Related to Healthy Body Index Revealed by Intestinal Microbiome and Metabolomics
Indian Journal of Microbiology (2022)
-
Non-alcoholic fatty liver diseases: from role of gut microbiota to microbial-based therapies
European Journal of Clinical Microbiology & Infectious Diseases (2020)
-
Oral Administration of Compound Probiotics Ameliorates HFD-Induced Gut Microbe Dysbiosis and Chronic Metabolic Inflammation via the G Protein-Coupled Receptor 43 in Non-alcoholic Fatty Liver Disease Rats
Probiotics and Antimicrobial Proteins (2019)
-
Hepatic Steatosis—a complex interaction of germs, genes and grub.
Pediatric Research (2018)
