
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are significant causes of morbidity and mortality in children and young adults. ADPKD, with an incidence of 1:400 to 1:1,000, affects more than 13 million individuals worldwide and is a major cause of end-stage renal disease in adults. However, symptomatic disease is increasingly recognized in children. ARPKD is a dual-organ hepatorenal disease with an incidence of 1:20,000 to 1:40,000 and a heterozygote carrier rate of 1 in 70. Currently, no clinically significant disease-specific therapy exists for ADPKD or ARPKD. The genetic basis of both ADPKD and ARPKD have been identified, and delineation of the basic molecular and cellular pathophysiology has led to the discovery that abnormal ADPKD and ARPKD gene products interact to create “polycystin complexes” located at multiple sites within affected cells. The extracellular matrix and vessels produce a variety of soluble factors that affect the biology of adjacent cells in many dynamic ways. This review will focus on the molecular and cellular bases of the abnormal cystic phenotype and discuss the clinical translation of such basic data into new therapies that promise to alter the natural history of disease for children with genetic PKDs.
Similar content being viewed by others


Epidemiology/Genetics/Clinical Features
The genetic polycystic kidney diseases of childhood (reviewed herein) are autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD). ADPKD (OMIM 173900; 613095) is the most common inherited human renal disease. Approximately 85% of ADPKD cases are caused by mutations in PKD1 on chromosome 16, and 15% are caused by mutations in PKD2 on chromosome 4. ADPKD is generally a late-onset, systemic disease characterized by bilateral, progressive enlargement of focal fluid-filled cysts occurring in all nephron segments with variable extrarenal manifestations. Extrarenal manifestations include cystic lesions in the liver, pancreas, spleen, and seminal vesicles; vascular abnormalities, including intracranial aneurysms, dilatation of the aortic root, and dissection of the thoracic aorta; mitral valve prolapse; abdominal and inguinal hernias; and early-onset hypertension (1).
Cysts in ADPKD kidneys form in utero (2,3) and clinical manifestations are increasingly being recognized in newborns, children, and adolescents. These include left ventricular hypertrophy with or without systemic hypertension, proteinuria, hematuria, nephrolithiasis, flank pain, and impaired renal function (3,4,5). A recent study of 52 consecutively identified pediatric ADPKD patients (mean age: 10 ± 4 y; range: 1–17) confirmed the presence of clinically relevant renal manifestations of ADPKD. Hypertension, albuminuria, or renal insufficiency was present in 77% of these patients (6).
The clinical spectrum of pediatric ADPKD ranges from a rare, severe neonatal presentation indistinguishable from ARPKD, to symptomatic disease noted above, to asymptomatic unilateral or bilateral renal cysts in normal or enlarged kidneys detected by ultrasonography, which remain clinically silent well into adulthood. Although all races and both sexes are equally affected, in adults, the renal phenotype may be severer in males, with liver manifestations severer in females. ADPKD affects ~600,000 individuals in the United States and 13 million individuals worldwide and accounts for 4–6% of the end-stage renal disease population in the United States and more than $1.5 billion in costs per year (7).
Recent studies in experimental models (8) and molecular screening of two consanguineous families indicate that some mutated alleles retain partial activity (hypomorphs) (9). These and other data demonstrate that the level of gene expression, presence of modifying genes, or untranslated messenger RNAs (mRNAs; i.e., microRNA) may also play a significant role in disease expression and progression of ADPKD (10).
ARPKD (OMIM 263200) belongs to a group of congenital hepatorenal fibrocystic syndromes and is a cause of significant renal and liver-related morbidity and mortality in children. ARPKD occurs less frequently (1:20,000 live births) than ADPKD, is commonly diagnosed in utero or at birth, and occurs as a result of mutations in a single gene, polycystic kidney and hepatic disease 1 (PKHD1) (11,12). The carrier status is estimated at 1 in 70 (13). Severely affected fetuses display a “Potter-like” oligohydramnios phenotype with lethal pulmonary hypoplasia and massively enlarged echogenic kidneys, which can compromise normal delivery. Improved neonatal and pediatric nephrology intensive care, in addition to enhanced disease recognition, have increased survival of the newborns, but death still occurs in 25–30% of affected neonates due to respiratory insufficiency (14,15).
Nearly 50% of affected individuals surviving the neonatal period progress to end-stage renal disease within the first decade of life (14,16). Liver disease is an invariable feature of ARPKD, and in some cases, it can be a predominant clinical feature. Of all ARPKD neonatal survivors, it is estimated that 40% have severe dual-organ disease, with the remainder equally divided into those with severe kidney/mild hepatobiliary disease, mild kidney/severe hepatobiliary disease, and mild dual-organ involvement (13,17,18). The 15-y survival of patients surviving the first year of life is projected to be >85% (5,15). Despite improved survival, morbidity of this dual-organ disease is significant due to the following reasons: (i) renal collecting duct ectatic cysts and marked renal enlargement, leading to hypertension and progressive renal failure and (ii) biliary dysgenesis, leading to abnormal bile duct formation with progressive periportal congenital hepatic fibrosis. Patients with congenital hepatic fibrosis develop portal hypertension with resulting esophageal or gastric varices, enlarged hemorrhoids, splenomegaly, hypersplenism, protein-losing enteropathy, and gastrointestinal bleeding (15). In addition to congenital hepatic fibrosis, nonobstructed dilatation of the intrahepatic bile ducts (Caroli’s syndrome) and dilatation of the common bile duct occur in more than 30% of ARPKD patients. The resultant abnormal hepatobiliary drainage contributes to a significant risk of ascending cholangitis with sepsis. Further, cholestasis may lead to malabsorption of fat-soluble vitamins (A, D, E, and K), and the overall abnormal proliferation of biliary cells has reportedly led to benign or malignant tumors in older patients. Management of portal hypertension may require endoscopic band ligation of esophageal varices or portosystemic shunting. In the severest instances of intractable portal hypertension, or severe dual renal and hepatobiliary disease, hepatic or dual renal–hepatic transplantation has become a viable option (18,19,20).
Mutation analysis of the PKHD1 gene has shown that ARPKD may present in individuals older than age 20, a finding that has significantly broadened the clinical spectrum of the disease (21). A recent report describes a 77-y-old asymptomatic patient who died with autopsy findings diagnostic of ARPKD/congenital hepatic fibrosis (22).
Cellular Pathophysiology and Translational Implications
The gene product of PKD1, polycystin-1 (PC1), is a 467-kDa receptor-like integral membrane protein containing a long N-terminal extracellular domain (23). It is predicted to be involved in cell–cell and cell–matrix interactions and also plays a role in calcium homeostasis through its physical interaction with polycystin-2, the protein product of PKD2 (24). PC1 interacts with the PKD2 gene product, polycystin-2 (PC2), via a short cytoplasmic C-terminus (25) to form a heterodimeric complex. PC1 is expressed throughout the body, including abdominal organs (kidney, liver, and pancreas), heart, and vasculature.
PKD2 encodes a much smaller, 110-kDa, 968-amino acid polypeptide, PC2) (26). PC2, also called transient receptor potential (TRP) P2, is a calcium-permeable, nonselective cation channel (27) that increases membrane permeability to Ca2+ and plays an essential role in polycystin-1 localization and function (28). PKD2 is generally a milder clinical disease than PKD1 due to the in utero development of fewer cysts in PKD2 rather than differences in the rate of cyst growth (29).
The ARPKD disease gene, PKHD1, encodes a 67-exon mRNA and is predicted to encode a protein of 4,074 amino acids called fibrocystin or polyductin (11,12). Fibrocystin is a novel integral membrane protein with an extensive, highly glycosylated N-terminal extracellular region, a single transmembrane domain, and a short cytoplasmic tail containing potential phosphorylation sites, a Ca2+-binding site, and a coiled coil domain that is responsible for numerous protein–protein interactions (12).
Despite the identification of the causative genes responsible for ADPKD and ARPKD, the precise function of these genes and their protein products is still unclear. This is due in part to many novel attributes of the genes: (i) the complexity of their protein structures, (ii) the large sizes of PKD1 and PKHD1, (iii) the multiple transcripts produced by these genes, (iv) the multiple intracellular sites of the PKD proteins, and (v) the participation of these proteins in a number of multimeric protein complexes. The lack of functional PC1, PC2, or fibrocystin is embryonically lethal, indicating that these proteins are essential and that PKD begins in utero (30). Studies from multiple laboratories and multiple models demonstrate that ADPKD and ARPKD share common phenotypic abnormalities and intersecting signaling pathways whose disruption leads to cyst formation (5,15,28,31).
Multiple studies have demonstrated that the protein products of the PKD1, PKD2, and PKHD1 genes, in addition to other proteins involved in renal cyst formation, exist in multimeric protein complexes (15,25,32,33,34). These large multimeric “cystoprotein complexes” normally exist at specific intracellular locations within renal epithelia. These sites include the apical cell membrane (particularly in, on, or adjacent to the primary cilium), adherens junctions, desmosomes, and focal adhesions. These protein complexes interact with multiple signaling pathways that are critical in maintaining normal tubular growth and differentiation. Functional changes in the structure and function of these complexes can occur if any of the individual protein components are altered. Mutations that modify cystoprotein complexes ultimately result in aberrant integration of complex signaling events and PKD.
Cyst initiation and expansion is a complex process characterized by abnormalities in tubular cell proliferation, fluid secretion, extracellular matrix formation, and cell polarity of specific proteins (35). It is also characterized by an imbalance between cellular proliferation and apoptosis. These fundamental processes are unbalanced in cystic kidneys.
Despite the lack of understanding of the precise molecular mechanisms by which mutations in PKD1, PKD2, and PKHD1 result in renal cyst formation, a number of common phenotypic abnormalities have been identified. These common phenotypic abnormalities define key pathogenic features of a unique phenotype of a cystic renal epithelial cell (5,15). The unique “cystic phenotype” is characterized by:
-
1
Quantitative and qualitative abnormalities in expression and function of the epidermal growth factor (EGF) family of receptors and ligands (EGF receptor (EGFR) axis);
-
2
Aberrant adenosine 3′,5′-cyclic monophosphate (cAMP) signaling, resulting in proliferation and decreased intracellular calcium;
-
3
Abnormal activity of C-terminal Src kinase or cellular Src (cSrc);
-
4
Abnormal structure and/or function of the primary cilia;
-
5
Alterations in planar cell polarity;
-
6
Alterations in cell–cell and cell–matrix interactions
The first three of the phenotypic features noted above are clearly mechanistic, whereas the others appear to be associative. Furthermore, the mechanistic abnormalities are those most likely to be amenable to translational pharmacological therapy and are therefore discussed in further detail.
Detailed Analysis of the Unique Phenotype of the Cystic Epithelial Cell
Mechanistic features of the cellular phenotype lead to the characteristic pathological features of cystic epithelia: a switch from well-differentiated, nonproliferative, resorptive epithelia to partially dedifferentiated, secretory epithelia characterized by specific polarization defects and high rates of proliferation and apoptosis (15,33,36). This is a critical functional change because mathematical modeling of renal epithelia determined that proliferation and secretion are necessary and sufficient to account for cyst growth in PKD (37).
EGFR-Axis-Mediated Proliferation and Secretion
Despite the complexities of the pathogenesis of cyst formation, cystic epithelia share common phenotypic abnormalities in spite of the different genetic mutations that underlie the diseases (15). Evidence from multiple laboratories demonstrate a significant role for abnormalities in the EGFR axis in promoting epithelial hyperplasia and ensuing renal cyst formation and progressive enlargement in both murine and human ADPKD and ARPKD (38,39,40,41). This is shown schematically in Figure 1 . Renal cystic epithelium demonstrates both quantitative (overexpression) and qualitative (mislocalization) of one or more members of the EGFR family of receptors (EGFR1, ErbB2, and ErbB4) and EGF-related ligands (EGFR axis or ErbB axis). Overexpression includes increased mRNA, increased protein, and increased activity or phosphorylation of the receptor, resulting in activation of multiple signaling pathways including the mitogen-activated protein kinase (MAPK) )pathway leading to increased proliferation (13,15). In addition, evidence from rodent models suggests that similar abnormalities of the EGFR axis may mediate the biliary epithelial hyperplasia and biliary ductal ectasia in ARPKD (41,42,43,44).

An overview of the epidermal growth factor receptor (EGFR) axis pathway that contributes to cyst formation and progressive enlargement in autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD). Abnormal expression and apical localization of the EGF receptors in the presence of increased ligand availability due to increased activity of matrix metalloproteinase (MMP) establishes an autocrine/paracrine cycle that drives cellular proliferation. Ligand binding leads to the formation of homodimers or heterodimers, which initiates autophosphorylation and activates the mitogen-activated protein kinase (MAPK) pathway, resulting in proliferation.
In addition, cyst fluid of ADPKD patients contains mitogenic concentrations of EGFR axis ligands, which are secreted into the lumen of cysts in amounts that induce cellular proliferation (45,46,47,48). In human ADPKD and ARPKD and in all animal models studied to date, abnormalities in the expression and localization of members of the EGFR axis have been reported. The apically expressed EGFR in cystic renal epithelia have been shown to specifically bind EGF/transforming growth factor-α with high affinity, autophosphorylate, and generate mitogenic signals in response to EGF and/or cyst fluid in vitro (40).
A role for increased EGFR tyrosine kinase activity in the formation and growth of renal cysts in vivo was firmly established by directly correlating cyst formation and growth with EGFR activity (phosphorylation levels). EGFR-specific tyrosine kinase inhibitors—either alone (44) or in combination with inhibitors of ligand availability (41,49)—demonstrated a direct correlation between reduced cyst formation and decreased EGFR activity (autophosphorylation).
Genetic complementation proved the mechanistic role of the abnormal EGFR axis phenotype in cystic disease. It was achieved by introducing a mutant EGFR with decreased tyrosine kinase activity, the wa2 mouse (50), via breeding into a murine model of ARPKD (39,51). This complementation demonstrated that the mutated form of the EGFR could block the increase in EGFR-specific tyrosine kinase activity that normally accompanies the development of renal cysts in PKD and it correlated directly with an improvement in kidney function and a substantial decrease in cyst formation in the renal collecting ducts. These data unequivocally establish a primary mechanistic role for the overexpression of the tyrosine kinase activity of the EGFR in the pathophysiology of renal cyst formation.
Similar in vivo therapeutic targeting of substrates, such as mitogen-activated protein kinases (the Ras-Raf-MEK-ERK pathway), downstream from the EGFR was effective in ameliorating biliary ductal cysts of the orthologous Pkhd1 PCK rat (43). Such data demonstrate analogous roles of abnormalities of the EGFR axis in both biliary and renal phenotypes of ARPKD.
Because renal cyst fluid contains biologically active EGF and transforming growth factor-α(52,53), it is possible that the increased EGFR tyrosine kinase activity in the collecting tubule is part of an autocrine/paracrine cycle that drives the cellular proliferation required for cyst growth (54). By analogy, one might suspect similar EGFR–ligand abnormalities in ARPKD biliary secretions. This is currently a focus of investigation in our laboratory.
In vitro studies using PKHD1-silenced HEK-293 T cell lines demonstrated that these cells were hyperproliferative on EGF stimulation, which lowered intracellular [Ca2+]i and caused EGF-induced increased ERK1/2 activity (55). An increase of intracellular [Ca2+]i in PKHD1-silenced cells repressed the EGF-dependent ERK1/2 activation and the hyperproliferative response to EGF stimulation. Thus, inhibition of PKHD1 can cause EGF-induced excessive proliferation through decreasing intracellular calcium concentration, [Ca2+]i, resulting in EGF-induced ERK1/2 activation. These studies suggest that loss of fibrocystin tissue leads to abnormal proliferation in kidney epithelial cells and cyst formation in ARPKD via modulation of intracellular [Ca2+] levels. As noted below, decreased [Ca2+]i is also a key feature of fluid secretion (56,57).
Multiple studies have demonstrated that collecting duct cells elevate [Ca2+] in response to flow across the cell surface, and this response has been attributed to PC1 and PC2 interactions in the primary cilia (58,59). However, intercalated cells, which lack primary cilia, respond to mechanical flow changes with increases in [Ca2+]i comparable with that observed in principal cells, which have primary cilia (60,61). Other TRP channels have been identified in collecting duct cells, and TRPV4 channels have been shown to respond to mechanical stimulation (62). Additionally, TRPV4 channels are essential for flow-induced [Ca2+] even in the presence of PKD2, suggesting that TRPV4 channels are responsible for the mechanosensitive properties of collecting duct cells (63,64).
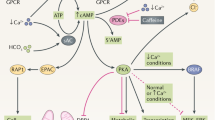
cAMP-Mediated Proliferation and Secretion
A second major contributor to renal epithelial cellular proliferation and secretion in both ADPKD and ARPKD is the adenylyl cyclase–cAMP pathway shown in Figure 2 . G protein–coupled vasopressin V2 receptors in the renal collecting duct regulate aquaporin 2 water channels and the activity of adenylyl cyclases, which in turn play a role in mediating intracellular cAMP levels.

A simplified representation of the cAMP or G-protein pathway that contributes to cyst formation and progressive enlargement in polycystic kidney disease (PKD). Adenylyl cyclase activation leads to increased levels of cAMP, which activates protein kinase A (PKA). PKA activation in the presence of low intracellular calcium levels phosphorylates β-Raf in an Src-dependent process. This phosphorylated β-Raf allows the cell to bypass the normal inhibition of PKA on mitogen-activated protein kinase activation and leads to increased cellular proliferation and possibly angiogenesis via activation of Erk 1/2.
In normal human and mouse renal epithelial cells, cAMP has been shown to inhibit the Ras/Raf-1/MEK/ERK pathway at the level of Raf-1 and to inhibit cell proliferation. In contrast, cAMP has been shown to stimulate β-Raf (via cSrc–dependent phosphorylation) and to activate the MEK/ERK pathway in ARPKD and ADPKD cells, leading to increased cell proliferation (65,66,67,68,69). Therefore, cAMP can act to stimulate or inhibit proliferation depending on intracellular conditions. This phenotypic shift in response to cAMP appears to involve cross talk between cAMP and Ca2+ and the cSrc signaling in response to β-Raf, a kinase upstream of the MEK/ERK pathway (57,70). In normal cells, β-Raf is repressed by Akt (protein kinase B), a Ca2+-dependent kinase, preventing cAMP activation of ERK and cell proliferation. In PKD epithelia from either ARPKD or ADPKD kidneys, low Ca2+ levels and cSrc-dependent phosphorylation of β-Raf result in an increased expression of cAMP and a switch in cAMP from an antiproliferative signaling molecule to a promitogenic stimulus that both activates the MEK/ERK pathway and promotes cellular proliferation and secretion (57,70).
The switch in cAMP from an antimitogenic to a mitogenic stimulus has been shown to be directly correlated with intracellular [Ca2]i (68). Pharmacological elevation of Ca2+ increases phospho-Akt levels, whereas Ca2+ channel blockers decrease phospho-Akt (69). This observation that the reduction of intracellular [Ca2]i in M-1 or normal human kidney cells with calcium channel blockers replicates the abnormal proliferative response of PKD cells to cAMP establishes a link among the PKD proliferative phenotype, reduction in [Ca2+]i, and cAMP activity (70).
Evidence for the role of [Ca2+]i-mediated proliferation has been further strengthened by studies showing that the elevation of [Ca2+]i levels in ADPKD-cultured and ARPKD-cultured cells increases Akt activity and blocks cAMP-dependent β-Raf and ERK activation (69). The study has also demonstrated that increases in [Ca2+]i restore the normal antimitogenic response to cAMP in renal cells derived from human ADPKD or ARPKD kidneys.
These data suggest that the PKD proteins (PC1, PC2, and fibrocystin) play a role in maintaining Ca2+ homeostasis. Mutations in any of the PKD proteins may lead to reduced [Ca2+]i and activate the cAMP mitogenic pathway.
Mechanisms of Fluid Secretion: A Key Feature Differentiating ADPKD From ARPKD
ARPKD cysts have a fundamental anatomical difference from ADPKD cysts, which would suggest different secretory mechanisms. Unlike ADPKD cysts, ARPKD cysts remain open to the urinary lumen, maintaining both afferent and efferent tubular connections, whereas ADPKD cysts rapidly “pinch off” from urinary flow and continue to expand by transepithelial secretion, as shown in Figures 1 and 2 . In ADPKD, more than 70% of cysts have no afferent or efferent tubular connections (71,72). Despite this anatomic difference, secretion is still a necessary element of cyst formation in both ADPKD and ARPKD. Given the structural differences and their continuity with urinary luminal flow, one would not expect transtubular secretion to be as critical in the pathogenesis of ARPKD cysts.
In ADPKD, the evidence indicates that Cl− is secreted via a cAMP-mediated cotransport mechanism in the basolateral membrane and via the cystic fibrosis transmembrane conductance regulator (CFTR) in the apical membrane, leading to expansion of cysts particularly after they have detached from the nephron of origin (57,70,73,74). This Cl− transport mechanism does not appear to be relevant in ARPKD. Using a genetic complementation approach, the BPK mouse was crossed with a CFTR knockout mouse (75). The results demonstrated that the absence of CFTR did not alter the course of renal or biliary cyst development or growth in ARPKD. Indeed, the double mutant appeared, if anything, to be larger and more cystic than cystic controls (76).
ARPKD cystic epithelia have been shown to exhibit net fluid secretion through a decrease in principal cell sodium absorption. This is due to an EGF-mediated decrease in the α subunit of the epithelial Na channel (77,78,79). However, conflicting data have been reported (80).
cSrc (pp66Src): Integration of EGFR Axis and cAMP-Mediated Proliferation and Secretory Pathways
Given the complex intracellular cascades activated in both ADPKD and ARPKD, the “holy grail” of future therapy is to identify key cellular “checkpoints” where diffuse processes responsible for the “cystic phenotype” are integrated. Given the primary roles of the EGFR axis and adenylyl cyclase–mediated proliferation, targeting “upstream” events that modulate multiple signaling pathways is a particularly intriguing approach.
cSrc (pp66Src) is a key focal intermediate in several central cystogenic signaling pathways, as shown in Figure 3 . Src is a key intermediate that integrates proliferation from both G-coupled protein receptor–cAMP pathway and the EGFR axis signaling pathways (5,57,70). Increased Src in PKD uncouples EGFR ligands from peritubular matrix, reciprocally phosphorylates EGFR, and is required for the phosphorylation of β-Raf, allowing a normal inhibitory mechanism and preventing renal epithelial proliferation to be bypassed. In animal models of PKD and in human PKD, increased Src activity (pY418) was found to be associated with a severer renal cystic disease. Furthermore, treatment of BPK mice and PCK rats with bosutinib, an inhibitor of Src, ameliorated both the renal and hepatic forms of the disease through inhibition of G-protein–coupled receptor and EGFR-axis-triggered phosphorylation cascades (31).

This represents our recent approach to the discovery and testing of potential therapeutic interventions. Our rationale is that by using a compound that inhibits both pathways, a smaller dose will be necessary to achieve maximum cyst reduction and any toxicity associated with the therapy will be minimized. cSrc provides such a target because it inhibits both the activity of the epidermal growth factor receptor (EGFR) axis and the G-protein (cAMP) pathway, which are both aberrantly active in autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD). Active cSrc can reciprocally phosphorylate EGFR and increase availability of EGFR axis ligands by activating matrix metalloproteinases (MMPs) necessary for ligand processing; furthermore, in the cAMP-mediated pathway, cSrc is necessary for phosphorylation of β-Raf and subsequent activation of mitogen-activated protein kinases (MAPK), leading to increased proliferation. This diagram demonstrates the rationale for the therapeutic strategy we are pursuing, the use of therapeutic compounds that interfere with multiple pathways.
From Pathophysiology to Current and Future Therapies
Preimplantation Genetic Diagnosis/In Vitro Fertilization
A novel, reliable linkage-based protocol for preimplantation genetic diagnosis of ARPKD using single-cell multiple-displacement amplification products for PKHD1 haplotyping has been developed, which significantly decreases the problem of allelic dropout (81). This unique protocol uses whole-genome amplification of single blastomeres, multiple-displacement amplification, and haplotype analysis with 20 novel polymorphic short tandem repeat markers from the PKHD1 gene and flanking sequences. This method enables unambiguous identification of the haplotypes of embryos produced by at-risk couples and eliminates the genetic risk (25%) of proven ARPKD carriers from having a child affected with ARPKD (81).
A similar approach to ADPKD has been published. However, the fact that many patients with ADPKD are relatively asymptomatic into adulthood raises major ethical concerns regarding this approach (82). Details regarding molecular diagnosis of ARPKD or ADPKD can be found at Gene Clinics: Clinical Genetic Information Resource (online database) at http://www.geneclinics.org.
Dialysis and Transplantation
Current therapy for ADPKD and ARPKD patients who have reached end-stage renal disease is limited to dialysis and transplantation. The treatment of dual-organ involvement in ARPKD has traditionally focused first on the renal complications and sequentially on hepatobiliary complications. In many cases, this approach leads to the necessity of a renal transplantation and a second transplant to deal with the severe hepatobiliary disease. A recently published article asserts that carefully chosen ARPKD patients with severe dual-organ involvement (40% of all neonatal survivors, as previously noted) who ultimately require kidney transplantation would potentially benefit from dual-organ (kidney and liver) transplant (18).
The authors reason that the complications of hepatic involvement (predominantly the risk of ascending cholangitis) (19), coupled with improved outcomes of liver transplantation, mandate a careful assessment of risk–benefit ratio in all ARPKD patients with severe dual-organ involvement. The authors assert that lower pediatric end-stage liver disease/model for end-stage liver disease scores can be used to justify the benefit of combined liver–kidney transplant compared with isolated kidney transplants. The authors present a step-by-step algorithm designed to assist clinicians in this decision-making process ( Figure 4 ) (18,19). A recent series of eight ARPKD patients receiving dual renal–hepatic transplantation demonstrate the feasibility of this approach (20). In January 2014, it seems appropriate to consider such an approach in selected patients, at least until the pharmacological, disease-specific options noted in the following section are proven safe and efficacious for ARPKD.

Algorithm to help guide clinicians in making decisions regarding performing an isolated kidney transplant vs. a combined liver–kidney transplant in autosomal recessive polycystic kidney disease/congenital hepatic fibrosis patients with dual-organ involvement. Reprinted with permission from (18). MELD, model for end-stage liver disease; PELD, pediatric end-stage liver disease.
The delineation of the molecular and cellular pathophysiology of PKD has led to a new era of pharmacological therapeutic intervention based on targeting of specific signaling abnormalities of cystic epithelia. The goal of these therapies is to prevent development or decrease the progressive enlargement of cystic lesions. The major targets or strategies currently being evaluated and our assessment of their potential (based on currently available data from ongoing human trials or preclinical considerations) are noted in Table 1 .
Table 1 broadly summarizes pharmacological approaches that have shown significant benefit in animal models of PKD and are either currently or soon will be tested for safety and efficacy in clinical trials. It is clear that all of the therapies listed have systemic side effects. The goal of clinical trials will be to determine safe pharmacological windows where efficacy is preserved and side effects minimized.
Based on systemic toxicities and preclinical studies to date, no single drug alone would appear to be fully capable of slowing the progressive renal cyst growth and decreased renal function that occur in ADPKD and ARPKD nor the biliary dysgenesis that leads to congenital hepatic fibrosis in ARPKD. Based on previously noted preclinical data (where the use of multiple agents reduced potential drug toxicity by 67% without a reduction in therapeutic efficacy) (41), future trials will most likely consider the use of the following: 1. multiple, complementary agents; or 2. agents that inhibit key intersecting points of abnormally active pathways (i.e., cSrc); or 3. inhibit multiple pathways using agents such as multikinase inhibitors.
Statement of Financial Support
This work was supported by National Institutes of Health grant DK079306 and the Children’s Research Institute of Children’s Hospital and Health System of Wisconsin.
Disclosure: The authors have no potential or perceived conflicts of interest.
References
Torres VE, Harris PC . Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med 2007;261:17–31.
Chapman AB, Guay-Woodford LM . Renal volume in children with ADPKD: size matters. Clin J Am Soc Nephrol 2009;4:698–9.
Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW . Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int 2008;74:1192–6.
Ecder T, Schrier RW . Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol 2009;5:221–8.
Sweeney WE Jr, Avner ED . Diagnosis and management of childhood polycystic kidney disease. Pediatr Nephrol 2011;26:675–92.
Selistre L, de Souza V, Ranchin B, Hadj-Aissa A, Cochat P, Dubourg L . Early renal abnormalities in children with postnatally diagnosed autosomal dominant polycystic kidney disease. Pediatr Nephrol 2012;27:1589–93.
Harris PC, Torres VE . Polycystic kidney disease. Annu Rev Med 2009;60:321–37.
Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 2004;13:3069–77.
Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int 2009;75:848–55.
Chandrasekaran K, Karolina DS, Sepramaniam S, et al. Role of microRNAs in kidney homeostasis and disease. Kidney Int 2012;81:617–27.
Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet 2002;70:1305–17.
Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 2002;30:259–69.
Dell KMR, Avner ED . Polycystic kidney disease, autosomal recessive. 2001 Jul 19. In: Pagon RA, Adam MP, Bird TD, et al, eds. Gene Reviews [online]. Seattle, WA: University of Washington, 1993–2013. http://www.ncbi.nlm.nih.gov/books/NBK1326/. http://www.geneclinics.org. doi (Accessed 22 September 2011).
Roy S, Dillon MJ, Trompeter RS, Barratt TM . Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol 1997;11:302–6.
Dell KM, Sweeney WE, Avner ED . Polycystic kidney disease. In: Avner ED, Harmon W, Niadet P, Yoshikawa N, eds. Pediatric Nephrology. 6th edn. Heidelberg, Berlin, Germany: Springer-Verlag, 2009: 849–88.
Kaplan BS, Kaplan P, Rosenberg HK, Lamothe E, Rosenblatt DS . Polycystic kidney diseases in childhood. J Pediatr 1989;115:867–80.
Gunay-Aygun M, Font-Montgomery E, Lukose L, et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 2013;144:112–121.e2.
Telega G, Cronin D, Avner ED . New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatr Transplant 2013;17:328–35.
Chapal M, Debout A, Dufay A, et al. Kidney and liver transplantation in patients with autosomal recessive polycystic kidney disease: a multicentric study. Nephrol Dial Transplant 2012;27:2083–8.
Brinkert F, Lehnhardt A, Montoya C, et al. Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): indication and outcome. Transpl Int 2013;26:640–50.
Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore) 2006;85:1–21.
Taneda S, Honda K, Aoki A, et al. An autopsy case of clinically un-diagnosed autosomal recessive polycystic kidney disease in 77-year-old male. Pathol Int 2012;62:811–6.
Consortium TIPKD. Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell 1995;81:289–98.
Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G . Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci USA 1997;94:6965–70.
Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG . PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 1997;16:179–83.
Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996;272:1339–42.
Koulen P, Cai Y, Geng L, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 2002;4:191–7.
Ong AC, Harris PC . Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int 2005;67:1234–47.
Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006;17:3013–9.
Chapman AB . The fetal environment: a critical phase that determines future renal outcomes in autosomal dominant polycystic kidney disease. Kidney Int 2012;81:814–5.
Sweeney WE Jr, von Vigier RO, Frost P, Avner ED . Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol 2008;19:1331–41.
Roitbak T, Ward CJ, Harris PC, Bacallao R, Ness SA, Wandinger-Ness A . A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol Biol Cell 2004;15:1334–46.
Wilson PD . Polycystic kidney disease. N Engl J Med 2004;350:151–64.
Ward CJ, Yuan D, Masyuk TV, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 2003;12:2703–10.
Chang MY, Ong AC . Mechanism-based therapeutics for autosomal dominant polycystic kidney disease: recent progress and future prospects. Nephron Clin Pract 2012;120:c25–34; discussion c35.
Harris PC, Rossetti S . Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab 2004;81:75–85.
Welling LW, Grantham JJ . Cystic and development diseases of the kidney. In: Brenner BM, Rector FC, eds. The Kidney. Philadelphia, PA: WB Saunders, 1991: 1657–94.
Du J, Wilson PD . Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol 1995;269(2 Pt 1):C487–95.
Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED . Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Invest 1998;101:935–9.
Sweeney WE Jr, Avner ED . Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am J Physiol 1998;275(3 Pt 2):F387–94.
Sweeney WE Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED . Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int 2003;64:1310–9.
Nauta J, Sweeney WE, Rutledge JC, Avner ED . Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr Res 1995;37:755–63.
Sato Y, Harada K, Kizawa K, et al. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am J Pathol 2005;166:49–60.
Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED . Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 2000;57:33–40.
Rohatgi R, Zavilowitz B, Vergara M, Woda C, Kim P, Satlin LM . Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr Nephrol 2005;20:552–3.
Sullivan LP, Wallace DP, Grantham JJ . Epithelial transport in polycystic kidney disease. Physiol Rev 1998;78:1165–91.
Ye M, Grant M, Sharma M, et al. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J Am Soc Nephrol 1992;3:984–94.
Sweeney WE, Avner ED . BPK cyst fluid contains EGF and TGF-a like peptides which are mitogenic and phosphorylate apical EGFR. J Amer Soc Nephrol 1996;7:1606.
Nemo R, Murcia N, Dell KM . Transforming growth factor alpha (TGF-alpha) and other targets of tumor necrosis factor-alpha converting enzyme (TACE) in murine polycystic kidney disease. Pediatr Res 2005;57(5 Pt 1):732–7.
Luetteke NC, Phillips HK, Qiu TH, et al. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev 1994;8:399–413.
Moyer JH, Lee-Tischler MJ, Kwon HY, et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994;264:1329–33.
Horikoshi S, Kubota S, Martin GR, Yamada Y, Klotman PE . Epidermal growth factor (EGF) expression in the congenital polycystic mouse kidney. Kidney Int 1991;39:57–62.
Ogborn MR, Sareen S . Transforming growth factor alpha and epidermal growth factor expression in experimental murine polycystic kidney disease. Pediatr Nephrol 1996;10:181–4.
Orellana SA, Avner ED . Cystic maldevelopment of the kidney. Semin Nephrol 1995;15:341–52.
Yang J, Zhang S, Zhou Q, et al. PKHD1 gene silencing may cause cell abnormal proliferation through modulation of intracellular calcium in autosomal recessive polycystic kidney disease. J Biochem Mol Biol 2007;40:467–74.
Pinto CS, Reif GA, Nivens E, White C, Wallace DP . Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am J Physiol Renal Physiol 2012;303:F1412–24.
Jansson K, Nguyen AN, Magenheimer BS, et al. Endogenous concentrations of ouabain act as a cofactor to stimulate fluid secretion and cyst growth of in vitro ADPKD models via cAMP and EGFR-Src-MEK pathways. Am J Physiol Renal Physiol 2012;303:F982–90.
Praetorius HA, Frokiaer J, Nielsen S, Spring KR . Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J Membr Biol 2003;191:193–200.
Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003;33:129–37.
Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM . Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 2003;285:F998–F1012.
Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O’Neil RG . Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J Biol Chem 2012;287:8782–91.
Wu L, Gao X, Brown RC, Heller S, O’Neil RG . Dual role of the TRPV4 channel as a sensor of flow and osmolality in renal epithelial cells. Am J Physiol Renal Physiol 2007;293:F1699–713.
Köttgen M, Buchholz B, Garcia-Gonzalez MA, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 2008;182:437–47.
Zaika O, Mamenko M, Berrout J, Boukelmoune N, O’Neil RG, Pochynyuk O . TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2013;24:604–16.
Yamaguchi T, Pelling JC, Ramaswamy NT, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 2000;57:1460–71.
Wallace DP, Christensen M, Reif G, et al. Electrolyte and fluid secretion by cultured human inner medullary collecting duct cells. Am J Physiol Renal Physiol 2002;283:F1337–50.
Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 2003;63:1983–94.
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP . Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004;279:40419–30.
Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP . Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 2006;17:178–87.
Wallace DP . Cyclic AMP-mediated cyst expansion. Biochim Biophys Acta 2011;1812:1291–300.
Grantham JJ, Geiser JL, Evan AP . Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int 1987;31:1145–52.
Ye M, Grantham JJ . The secretion of fluid by renal cysts from patients with autosomal dominant polycystic kidney disease. N Engl J Med 1993;329:310–3.
Belibi FA, Reif G, Wallace DP, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 2004;66:964–73.
Terryn S, Ho A, Beauwens R, Devuyst O . Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 2011;1812:1314–21.
Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC . Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science 1992;257:1125–8.
Nakanishi K, Sweeney WE Jr, Macrae Dell K, Cotton CU, Avner ED . Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2001;12:719–25.
Veizis IE, Cotton CU . Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells. Am J Physiol Renal Physiol 2005;288:F474–82.
Levchenko V, Zheleznova NN, Pavlov TS, Vandewalle A, Wilson PD, Staruschenko A . EGF and its related growth factors mediate sodium transport in mpkCCDc14 cells via ErbB2 (neu/HER-2) receptor. J Cell Physiol 2010;223:252–9.
Zheleznova NN, Wilson PD, Staruschenko A . Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim Biophys Acta 2011;1812:1301–13.
Rohatgi R, Greenberg A, Burrow CR, Wilson PD, Satlin LM . Na transport in autosomal recessive polycystic kidney disease (ARPKD) cyst lining epithelial cells. J Am Soc Nephrol 2003;14:827–36.
Lau EC, Janson MM, Roesler MR, Avner ED, Strawn EY, Bick DP . Birth of a healthy infant following preimplantation PKHD1 haplotyping for autosomal recessive polycystic kidney disease using multiple displacement amplification. J Assist Reprod Genet 2010;27:397–407.
De Rycke M, Georgiou I, Sermon K, et al. PGD for autosomal dominant polycystic kidney disease type 1. Mol Hum Reprod 2005;11:65–71.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sweeney, W., Avner, E. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res 75, 148–157 (2014). https://doi.org/10.1038/pr.2013.191
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2013.191
This article is cited by
-
Predictors of progression in autosomal dominant and autosomal recessive polycystic kidney disease
Pediatric Nephrology (2021)
-
Management of delivery of a fetus with autosomal recessive polycystic kidney disease: a case report of abdominal dystocia and review of the literature
Journal of Medical Case Reports (2019)
-
Aberrant Regulation of Notch3 Signaling Pathway in Polycystic Kidney Disease
Scientific Reports (2018)
-
Prenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases
Archives of Gynecology and Obstetrics (2017)
-
Genetic analysis of the PKHD1 gene with long-rang PCR sequencing
Journal of Huazhong University of Science and Technology [Medical Sciences] (2016)
