
Abstract
Premature infants are at increased risk for persistent growth failure, neurodevelopmental impairment, hypertension, and diabetes. Rapid neonatal growth has been linked to the increasing prevalence of diabetes and obesity. Nutritional goals for the premature infant with incipient growth failure have thus become a source of controversy. We used isogenic mice with natural variation in perinatal growth to test the hypothesis that neonatal catch up growth improves the neurobehavioral and cardiovascular outcomes of low-birth weight mice, despite an increase in diabetes susceptibility. Adult mice that experienced prenatal and neonatal growth restriction had persistent growth failure, hypertension, and neurobehavioral alterations. When switched from standard rodent chow to a hypercaloric diet, growth restricted mice were protected from diet-induced obesity. Among low-birth weight male mice, neonatal catch up growth normalized neurobehavioral and cardiovascular phenotypes, but led to insulin resistance and high fat diet-induced diabetes. Among low-birth weight female mice, neonatal catch up growth did not prevent the development of adult hypertension and significantly increased measures of anxiety, including self-injury and the avoidance of open spaces. These studies support the importance of the perinatal environment in the resetting of adult disease susceptibility and suggest an earlier window of vulnerability among growth restricted female mice.
Similar content being viewed by others


Main
Restricted perinatal growth is an independent risk factor for the development of type 2 diabetes, hypertension, and coronary heart disease (1,2). The primary animal models of programmed adult disease induce fetal growth restriction through maternal nutrient restriction or uterine artery ligation (3). The development of diabetes and obesity in these models has been most dramatic when rodent pups are cross-fostered to well-nourished dams and then weaned to postnatal high-fat diets (4,5). These studies are consistent with epidemiologic data linking excessive weight gain in infancy with adult metabolic syndrome (6). Although these data suggest that restriction of neonatal nutrition might improve adult metabolic health, a comprehensive approach is essential when considering the long-term consequences of perinatal dietary alterations (7).
Preterm delivery increases the risk of impaired growth during a critical window of brain development. By 36-wk postmenstrual age, ∼90% of premature infants have growth failure (weight less than the 10th percentile of reference fetuses) (8). This perinatal growth failure is associated with permanent reductions in adult weight and height (9). It follows that premature delivery greatly increases the risk of neurodevelopmental impairment, insulin resistance, and adult hypertension (10–12). As parallel epidemics of prematurity and obesity unfold, there is a critical need for research-based neonatal nutritional recommendations that seek to optimize short-term health and long-term outcomes.
We previously evaluated the physiologic consequences of natural perinatal growth restriction in isogenic mice (13). We sought to expand on this model by evaluating the long-term consequences of neonatal catch up growth and investigating the programming of diet-induced obesity. We hypothesized neonatal catch up growth would improve the neurobehavioral and cardiovascular outcomes of low-birth weight mice, but increase their susceptibility to high-fat diet-induced obesity.
METHODS
Animal model.
The investigation was approved by the University of Iowa Animal Care and Use Committee and conforms to National Institutes of Health guidelines. Adult C57Bl/6J mice (Jackson Laboratory, Bar Harbor, ME) were bred for 2 wk and then moved into individual cages. Dams delivered naturally, fostered their own pups, and were maintained on standard rodent chow (4 kcal/g, 6% of energy as fat; 7013; Harlan Teklad, Madison, WI) throughout pregnancy and lactation. Litters were never culled. To lessen the risk of rejection, nests were undisturbed for 24 h after delivery, and initial pup weights were obtained on postnatal d 2. From d 2 to 20, pups were marked with indelible ink and daily weights were recorded. Following World Health Organization guidelines, growth restriction was identified by a gender-specific weight below the 10th percentile (14). Fetal or neonatal growth restriction was, therefore, defined by a weight less than the 10th percentile for the colony on postnatal d 2 or 20, respectively. Appropriate growth was defined by a weight within one SD of the colony mean. Pups were weaned to individual cages and standard rodent chow on postnatal d 21. Mice were continuously recruited into this study from a birth cohort spanning from June 2007 to October 2007. Each study group consisted of mice from at least five different litters, with no more than two mice obtained from a given litter.
Baseline endocrine studies.
At 20 wk, feed intake was measured by recording initial feed weight and subtracting the amount remaining 3 wk later. Glucose and insulin tolerance tests were then performed after a 3-h fast, as previously described (13). For glucose tolerance tests, 20% dextrose was administered by intraperitoneal injection (2 g/kg). After at least 48 h of recovery, insulin tolerance tests were performed by intraperitoneal injection of regular human insulin (0.75 U/kg; Humulin-R, Eli Lilly, Indianapolis, IN).
Neurocardiovascular studies.
After the initial endocrine tests, tail cuff systolic blood pressure (SBP) and heart rate were recorded for 5 consecutive days, as previously described (13). Unconditioned anxiety was measured by placing mice in a 40.6 × 40.6 cm open field (San Diego Instruments, San Diego, California) (15). Infrared beam breaks were recorded for 5 min on 2 consecutive days. To further evaluate anxiety-associated thigmotaxis (tendency to remain along the chamber's walls), center activity was defined as infrared beam breaks occurring inside of a 7.62 cm border. Barnes maze testing was then used to assess spatial learning and memory. The Barnes maze exploits the instinct of small rodents to seek escape to a sheltered environment when placed in an open arena under aversive illumination. The maze consists of a circular platform (92 cm diameter) with 20 equidistant holes around the perimeter. One hole leads to an escape box that is consistently oriented with respect to visuo-spatial cues throughout the room. Each mouse was given four trials per day, with an inter-trial interval of 1 h. The initial training day was followed by four consecutive test days to evaluate spatial learning. Mice were retested 2 wk later to assess memory consolidation and long-term retention. A video camera recorded each session. Escape latency (time taken to find and enter the escape box) was recorded by observers blinded to group assignment.
High-fat diet.
After completion of the baseline studies, all mice were switched to a hypercaloric, high-fat diet (5 kcal/g, 45% of energy as fat; D12451; Research Diets, New Brunswick, NJ). The caloric composition of the diet included 45% fat (39% lard and 6% soybean oil), 35% carbohydrates (18% sucrose, 10% maltodextrin, and 7% corn starch), and 20% protein (19.7% casein and 0.3% l-cystine). During the first week of high-fat diet administration, an exercise wheel (35 cm circumference; Thoren Caging Systems, Hazleton, PA) was placed inside each cage to evaluate voluntary activity. Exercise wheel revolutions were digitally recorded over the ensuing 72 h. Twenty weeks later, glucose and insulin tolerance tests were repeated. Insulin levels were measured on plasma collected from fasting mice immediately before glucose tolerance testing, as previously described (13).
Indirect calorimetry.
While receiving the high-fat diet, mice were housed in individual, airtight chambers warmed to thermoneutrality (30°C) by a circulating water bath. Oxygen consumption (VO2) and carbon dioxide production (VCO2) were calculated based on the gas composition flowing into and away from the closed-circuit during the constant flow of air (AEI Technologies oxygen analyzer S-3A and carbon dioxide analyzer CD-3A). To approximate basal metabolic rate, VO2 was calculated at thermoneutrality, after mice had fallen asleep in the midst of the light cycle. RQ was calculated by dividing VCO2 by VO2.
Data analysis.
All values are presented as mean ± SE. Statistical comparisons were performed by 2-tailed t test or two-way ANOVA with Holm Sidak testing, as appropriate. A value of p < 0.05 was considered significant. All analyses were performed using SigmaStat 3.0 (SPSS Inc., Chicago, IL).
RESULTS
Establishment of the model.
Median litter size was seven pups with an interquartile range of six to nine pups. There were slightly more male than female pups on d 2 (53% male, p = 0.31 by χ2 analysis). Newborn mouse weights were normally distributed (Fig. 1A; Shapiro-Wilk test, W = 0.99). Female mice weighed significantly less than male mice on d 2 (female: 1.76 ± 0.02 g; male: 1.82 ± 0.02 g; p < 0.05) and d 20 (female: 8.1 ± 0.1 g; male: 8.4 ± 0.1 g; p < 0.05). Litter size influenced both d 2 pup weight (R = 0.80, p < 0.05) and neonatal weight gain (Fig. 1B; R = 0.94, p < 0.05). Based on the initial 178 male mice, growth restriction (weight below the 10th percentile) was defined by weights below 1.56 g on d 2 and below 7.1 g on day 20. Based on the initial 156 female mice, the corresponding 10th percentile weights were 1.50 g on d 2 and 6.8 g on d 20. Of the mice with fetal growth restriction, 50% of the males and 35% of the females achieved catch up growth.

Isogenic male ▪ and female ○ C57BL/6 mice had normally distributed birth weights (A, n = 334 mice, W = 0.99). Neonatal weight gain was inversely proportional to litter size (B, n = 57 litters, R = 0.94). Fetal or neonatal growth restriction was defined by a weight below the 10th percentile at 2 d (C) or 20 d (D). Using this definition, mice were categorized as having appropriate fetal and neonatal growth (AA, ▪, n = 15 males and 14 females), restricted fetal growth but appropriate neonatal growth (RA,  , n = 6 males and 8 females), or restricted fetal and neonatal growth (RR, □, n = 7 males and 6 females). *p < 0.01 vs control.
, n = 6 males and 8 females), or restricted fetal and neonatal growth (RR, □, n = 7 males and 6 females). *p < 0.01 vs control.
By definition, mice with combined fetal and neonatal growth restriction (RR mice) had significantly reduced weights on d 2 and 20. In contrast, mice with fetal growth restriction followed by accelerated neonatal growth (RA mice) had significant reductions in only d 2 weight (Fig. 1C and D). Based on the observed catch up rates and the definition of growth restriction (weight less than the 10th percentile), 5% of males were RR, 5% of males were RA, 6.5% of females were RR, and 3.5% of females were RA. Mice were continuously recruited until the final cohorts of RR and RA mice were established (six to eight mice per group). For every mouse recruited into the RR and RA groups, mice with appropriate fetal and neonatal growth (AA mice) were retained (15 males and 14 females). For most litters, AA pups were fostered along side RA and/or RR pups.
Adult size and caloric intake.
RR male and female mice had persistent growth failure (Fig. 2). The reductions in adult weight (decrease of 31% in males and 13% in females), length (decrease of 7% in males and 5% in females), and brain weight (decrease of 9% in males and 5% in females) were associated with a significant reduction in feed intake (kcal/d) for both male and female RR mice (Fig. 2D). Consistent with asymmetric growth restriction, brain weight normalized to body weight (mg/g) was significantly greater in adult male and female RR mice (RR male 18.2 ± 0.7 versus AA male 14.7 ± 0.8; RR female 21.0 ± 0.7 versus AA female 19.2 ± 0.4). When also corrected for current body weight, adult feed intake (expressed as kcal/g/d) was not influenced by perinatal growth restriction. RA and AA mice had similar weights, lengths, and caloric intakes. Female, but not male, RA mice had a significant reduction in adult brain weight (Fig. 2C).

Adult weight (A), length (B), brain weight (C), and caloric intake (D) were measured among AA (▪, n = 15 males and 14 females), RA (, n = 6 males and 8 females), and RR (□, n = 7 males and 6 females) mice. *p < 0.05 and **p < 0.01 vs AA.
Phenotypes on standard chow.
Although perinatal growth restriction did not alter fasting glucose levels or glucose tolerance at 4 mo, male RA mice had a significant reduction in insulin sensitivity (Fig. 3). Male and female RR mice, as well as female RA mice were hypertensive (Fig. 4A). Perinatal growth patterns did not significantly alter adult heart rates (Fig. 4B). Male RA mice had decreased activity when placed into an open field (Fig. 4C). Male and female RR mice were also less active in the open field, whereas female RA mice had skewed distribution of movement along the periphery of the field (Fig. 4D).

AA (▪, n = 15 males and 14 females), RA (, n = 6 males and 8 females), and RR (□, n = 7 males and 6 females) mice received standard feed for 20 wk. Glucose levels were then measured while fasting (A) for intraperitoneal glucose tolerance tests (B), or insulin tolerance tests (C, male; D, female). *p < 0.05 vs AA.

Tail cuff systolic blood pressure (A) and heart rate (B) were recorded among AA (▪, n = 15 males and 14 females), RA (, n = 6 males and 6 females), and RR (□, n = 7 males and 6 females) mice. To measure generalized anxiety, these mice were then placed into a 40.6 × 40.6 cm open-field and infrared beam breaks were recorded for 5 min (C). Beam breaks into the center were normalized by total activity (D). *p < 0.05 and **p < 0.01 vs AA.
After open-field testing, four of the eight female RA mice developed open sores from excessive grooming (an anxiety-linked behavior) and meaningful data could no longer be obtained from that study population. During Barnes maze training, male RR mice escaped from the aversive environment much faster than controls (Fig. 5A). This anxiety driven behavior hampered definitive testing of subsequent learning in male mice (Fig. 5B). Female RR mice had significantly impaired learning (Fig. 5C) and memory, evidence by their delayed escape from the Barnes maze 2 wk after training (Fig. 5D).

After open-field testing, four of the eight female RA mice developed open sores from excessive grooming, and meaningful data could no longer be obtained from that study population. All other AA (▪, n = 8 males and 9 females), RA (, n = 6 males), and RR (□, n = 6 males and 6 females) mice were placed onto a brightly illuminated Barnes maze and escape time was measured (A). Male (B) and female (C) mice were then trained for 5 consecutive days, and visuo-spatial memory was evaluated 14 d later (test day 19, D). *p < 0.05 and **p < 0.01 vs AA.
Phenotypes on high-fat chow.
Male mice with neonatal catch up growth (RA mice) had fasting hyperglycemia, hyperinsulinemia, glucose intolerance, and insulin resistance while receiving a high-fat diet (Fig. 6). Female mice with perinatal growth failure (RR mice) had significantly improved glucose tolerance (Fig. 6C) without significant alterations in insulin sensitivity (Fig. 6D).

After AA (▪, n = 14 males and 14 females), RA (, n = 6 males), and RR (□, n = 7 males and 6 females) mice were switched to high-fat feed for 20 wk, glucose (A) and insulin (B) levels were measured while fasting. Mice then received intraperitoneal glucose (2 g kg−1; C) or insulin (0.75 units kg−1; D, squares males and circles females) and glucose levels were monitored. *p < 0.05 vs AA.
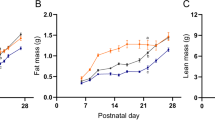
While on the high-fat diet, male and female RR mice gained significantly less weight than controls (Fig. 7A). In male RR mice, this protection from diet-induced obesity was associated with a significant increase in exercise and basal metabolic rate (resting VO2) (Fig. 7). Although male RA mice did not have excessive weight gain on a postnatal high-fat diet, they did have a reduction in voluntary exercise that was consistent with open field testing (Fig. 7B). Respiratory quotients were not significantly influenced by perinatal growth patterns (Fig. 7D).

Weight gain during the 20 wk of high-fat diet administration was measured among AA (▪, n = 14 males and 14 females), RA (, n = 6 males), and RR (□, n = 7 males and 6 females) mice (A). During the initial week of high-fat diet administration, exercise wheels were placed into the cages to record voluntary activity (B). By indirect calorimetry, resting VO2 (C), and RQ (D) were then determined. *p < 0.05 and **p < 0.01 vs AA.
DISCUSSION
Over the past decade, investigations into the developmental origins of adult disease have raised awareness of the multifaceted complications of an adverse intrauterine environment. Although demonstration that fetal growth restriction increases the risk of adult disease has prompted interest in the promotion of perinatal growth, studies have shown neonatal over-nutrition may exacerbate, rather than prevent, the evolution of adult disease (16,17). In contrast, studies in male mice have suggested neonatal malnutrition may prolong lifespan and prevent the development of diet-induced obesity (18–20). We expanded upon these studies in demonstrating that natural perinatal growth restriction provides protection from diet-induced metabolic syndrome, but at the expense of neurocardiovascular health.
Mice with combined RR mice remained small as adults. This reduced adult size is consistent with data showing premature and small for gestational age term infants have permanent reductions in adult height and weight (9). Our data showing that perinatal growth restriction leads to adult hypertension are consistent with data from the Helsinki birth cohort showing hypertension strongly associates with impaired growth through the first 4 y of life (1). The blood pressure normalization we demonstrated following catch up growth in male mice differs from the findings of Singhal et al. (17), which showed that neonatal overnutrition increases diastolic blood pressure among low-birth weight term infants. Apart from potential species differences, comparisons are hampered by the limited caloric intake data obtained in our study, the clinical trial's limited follow-up (51%) and a significantly enriched proportion of males in the clinical trial's overnutrition population. Further studies with adequate power to demonstrate sex-specific effects are needed to better define the role of neonatal catch up growth in the programming of hypertension.
Controlling for confounding variables, reduced neonatal brain growth has been consistently associated with long-term intellectual and behavioral impairment (21,22). Our data strengthen this association in showing genetically identical growth restricted mice raised in identical postnatal environments have permanent reduction in brain weight and cognitive performance. Interestingly, the decline in brain growth and cognitive performance among growth-restricted mice was strongly paralleled by the presence of tail cuff hypertension. Given the physical restraint and handling required with tail cuff blood pressure measurements, it is possible the observed hypertension is a further expression of programmed anxiety.
We used two separate behavioral tests to evaluate the presence of anxiety. Mice with perinatal growth restriction displayed markedly decreased movement when placed within an open field chamber. This behavior seemed to be related to anxiety, rather than a general reduction in movement, as growth restricted male mice had significantly increased activity on exercise wheels, and they escaped more rapidly from the stress-evoking Barnes maze. Related to these results, Vickers et al. (23) have shown both maternal undernutrition and postnatal overnutrition decrease adult rat movement in an anxiety-provoking behavioral apparatus. Further studies are necessary to evaluate the roles of fetal and postnatal growth on the programming of locomotor activity under a variety of environmental conditions.
Intriguingly, neonatal catch up growth normalized Barnes maze performance for male mice, whereas catch up growth accentuated the expression of anxiety in female mice. In concert with the brain weight data, these data suggest female mice have an earlier neurodevelopmental window of susceptibility, such that later interventions, including neonatal catch up growth, are less likely to modify female mouse neurocognitive performance. Considering the relative immaturity of the mouse brain at the time of delivery (roughly analogous to the developmental stage of the third trimester human fetus), these data are consistent with clinical data showing premature male infants are at twice the risk of growth failure-associated neurodevelopmental impairment (24). This sexually dimorphic neonatal window of vulnerability (or reversibility) may also underlie the greater reduction in adult size seen in male RR mice fostered in large litters, and the higher rate of postnatal catch up growth seen in male mice fostered in small litters. The etiologies for this sexual dimorphism in growth trajectory and resultant susceptibility to neurocardiovascular disease are not known.
Consistent with an expanding body of literature, catch up neonatal growth led to insulin resistance among low-birth weight male mice. These mice had marked insulin resistance without significant hyperglycemia on standard chow, suggesting that the loss in insulin sensitivity precedes the development of diet-induced hyperglycemia. This speculation is further supported by the 4-fold increase in fasting insulin among these mice, as well as clinical data showing that among premature infants, neonatal growth velocity is inversely associated with adult insulin sensitivity (6). With data supporting a role for early exercise in the prevention of insulin resistance and diabetes (25), the reduction in exercise seen among the diabetes-prone male RA mice merits further consideration as a target for early intervention.
Our data are consistent with previous findings in male offspring of undernourished rats (5,26). Maternal starvation to 30% of ad libitum intake throughout pregnancy reduced offspring weight through adulthood and programmed tail cuff hypertension and hyperinsulinemia (5). Similarly, profound maternal malnutrition (60% protein restriction) throughout pregnancy and lactation persistently decreased the body weight of adult offspring (26). As demonstrated in our study, postnatal hypercaloric nutrition amplified these programmed phenotypes. Intriguingly, both of these studies showed prenatal undernutrition followed by neonatal catch up growth significantly increases retroperitoneal fat pad weight in male rats, suggesting a possible mechanism for the insulin resistance we observed in RA male mice.
Although the degree of self-injury among female RA mice made definitive testing in this gender impossible, our baseline data support a greater predisposition for male mice to programmed diabetes. This finding is consistent with data supporting a sexually dimorphic susceptibility to diabetes among inbred C57BL/6 male mice (27,28), and enhanced diabetes susceptibility among protein malnourished male, but not female, rats (29).
Among the factors that regulate postnatal growth, we evaluated caloric intake, resting energy expenditure, and voluntary activity. Perinatal growth restriction seemed to reset a number of homeostatic mechanisms, including a reduction in caloric intake. In addition to this reduced intake, perinatal growth restricted male mice had increased exercise wheel utilization and increased resting energy expenditure. Although the perinatal origins of this altered resting energy expenditure have not been described, the trend seen toward increased heart rate might be contributory.
By allowing pups to wean naturally to standard rodent feed, our study uniquely mirrored the typical growth patterns seen in other naturally growth restricted populations. We demonstrated that naturally occurring perinatal growth restriction leads to hypertension and behavioral alterations but overrides genetic predisposition and protects the same mice from metabolic syndrome when a high-fat diet is introduced in adulthood. These findings support previous studies demonstrating that accelerated postnatal growth may be associated with detrimental effects on adult metabolic control, but postnatal growth impairment negatively influences neurocardiovascular outcomes. Further studies are needed to identify strategies that facilitate brain growth while simultaneously protecting the survivors of adverse intrauterine exposures from increasingly harsh postnatal environments. Identification of candidate genes and genetic polymorphisms holds the promise of tailored pharmacologic and nutritional interventions to better match disease susceptibilities, perinatal growth, and the prevailing environment.
Abbreviations
- AA:
-
appropriate fetal growth, appropriate neonatal growth
- RA:
-
restricted fetal growth, appropriate neonatal growth
- RR:
-
restricted fetal growth, restricted neonatal growth
References
Eriksson JG, Forsen TJ, Kajantie E, Osmond C, Barker DJ 2007 Childhood growth and hypertension in later life. Hypertension 49: 1415–1421
Eriksson JG, Osmond C, Kajantie E, Forsen TJ, Barker DJ 2006 Patterns of growth among children who later develop type 2 diabetes or its risk factors. Diabetologia 49: 2853–2858
McMillen IC, Robinson JS 2005 Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85: 571–633
Vickers MH, Reddy S, Ikenasio BA, Breier BH 2001 Dysregulation of the adipoinsular axis—a mechanism for the pathogenesis of hyperleptinemia and adipogenic diabetes induced by fetal programming. J Endocrinol 170: 323–332
Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD 2000 Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 279: E83–E87
Rotteveel J, van Weissenbruch MM, Twisk JW, Delemarre-Van de Waal HA 2008 Infant and childhood growth patterns, insulin sensitivity, and blood pressure in prematurely born young adults. Pediatrics 122: 313–321
Jimenez-Chillaron JC, Patti ME 2007 To catch up or not to catch up: is this the question? Lessons from animal models. Curr Opin Endocrinol Diabetes Obes 14: 23–29
Dusick AM, Poindexter BB, Ehrenkranz RA, Lemons JA 2003 Growth failure in the preterm infant: can we catch up?. Semin Perinatol 27: 302–310
Hack M, Schluchter M, Cartar L, Rahman M, Cuttler L, Borawski E 2003 Growth of very low birth weight infants to age 20 years. Pediatrics 112: e30–e38
Keijzer-Veen MG, Finken MJ, Nauta J, Dekker FW, Hille ET, Frölich M, Wit JM, van der Heijden AJ 2005 Is blood pressure increased 19 years after intrauterine growth restriction and preterm birth? A prospective follow-up study in The Netherlands. Pediatrics 116: 725–731
Hovi P, Andersson S, Eriksson JG, Järvenpää AL, Strang-Karlsson S, Mäkitie O, Kajantie E 2007 Glucose regulation in young adults with very low birth weight. N Engl J Med 356: 2053–2063
Casey PH, Whiteside-Mansell L, Barrett K, Bradley RH, Gargus R 2006 Impact of prenatal and/or postnatal growth problems in low birth weight preterm infants on school-age outcomes: an 8-year longitudinal evaluation. Pediatrics 118: 1078–1086
Roghair RD, Aldape G 2007 Naturally occurring perinatal growth restriction in mice programs cardiovascular and endocrine function in a sex- and strain-dependent manner. Pediatr Res 62: 399–404
1995 Physical status: the use and interpretation of anthropometry. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 854: 1–452.
Crawley JN 2008 Behavioral phenotyping strategies for mutant mice. Neuron 57: 809–818
Singhal A, Cole TJ, Fewtrell M, Deanfield J, Lucas A 2004 Is slower early growth beneficial for long-term cardiovascular health?. Circulation 109: 1108–1113
Singhal A, Cole TJ, Fewtrell M, Kennedy K, Stephenson T, Elias-Jones A, Lucas A 2007 Promotion of faster weight gain in infants born small for gestational age: is there an adverse effect on later blood pressure?. Circulation 115: 213–220
Ozanne SE, Hales CN 2004 Lifespan: catch-up growth and obesity in male mice. Nature 427: 411–412
Jimenez-Chillaron JC, Hernandez-Valencia M, Lightner A, Faucette RR, Reamer C, Przybyla R, Ruest S, Barry K, Otis JP, Patti ME 2006 Reductions in caloric intake and early postnatal growth prevent glucose intolerance and obesity associated with low birthweight. Diabetologia 49: 1974–1984
Ozanne SE, Lewis R, Jennings BJ, Hales CN 2004 Early programming of weight gain in mice prevents the induction of obesity by a highly palatable diet. Clin Sci (Lond) 106: 141–145
Gale CR, O'Callaghan FJ, Bredow M, Martyn CN 2006 The influence of head growth in fetal life, infancy, and childhood on intelligence at the ages of 4 and 8 years. Pediatrics 118: 1486–1492
Ehrenkranz RA, Dusick AM, Vohr BR, Wright LL, Wrage LA, Poole WK 2006 Growth in the neonatal intensive care unit influences neurodevelopmental and growth outcomes of extremely low birth weight infants. Pediatrics 117: 1253–1261
Vickers MH, Breier BH, McCarthy D, Gluckman PD 2003 Sedentary behavior during postnatal life is determined by the prenatal environment and exacerbated by postnatal hypercaloric nutrition. Am J Physiol Regul Integr Comp Physiol 285: R271–R273
Hintz SR, Kendrick DE, Vohr BR, Kenneth Poole W, Higgins RD 2006 Gender differences in neurodevelopmental outcomes among extremely preterm, extremely-low-birthweight infants. Acta Paediatr 95: 1239–1248
Patterson CM, Dunn-Meynell AA, Levin BE 2008 Three weeks of early-onset exercise prolongs obesity resistance in DIO rats after exercise cessation. Am J Physiol Regul Integr Comp Physiol 294: R290–R301
Bieswal F, Ahn MT, Reusens B, Holvoet P, Raes M, Rees WD, Remacle C 2006 The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity (Silver Spring) 14: 1330–1343
Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, Quarterman J, Freeman HC, Ashcroft FM, Cox RD 2005 A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48: 675–686
Knight BS, Pennell CE, Adamson SL, Lye SJ 2007 The impact of murine strain and sex on postnatal development after maternal dietary restriction during pregnancy. J Physiol 581: 873–881
Zambrano E, Bautista CJ, Deás M, Martínez-Samayoa PM, González-Zamorano M, Ledesma H, Morales J, Larrea F, Nathanielsz PW 2006 A low maternal protein diet during pregnancy and lactation has sex- and window of exposure-specific effects on offspring growth and food intake, glucose metabolism and serum leptin in the rat. J Physiol 571: 221–230
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by NIH grant HD050359 and funds from the Children's Miracle Network.
Rights and permissions
About this article
Cite this article
Hermann, G., Miller, R., Erkonen, G. et al. Neonatal Catch Up Growth Increases Diabetes Susceptibility But Improves Behavioral and Cardiovascular Outcomes of Low Birth Weight Male Mice. Pediatr Res 66, 53–58 (2009). https://doi.org/10.1203/PDR.0b013e3181a7c5fd
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181a7c5fd
This article is cited by
-
LRP6 Bidirectionally Regulates Insulin Sensitivity through Insulin Receptor and S6K Signaling in Rats with CG-IUGR
Current Medical Science (2023)
-
Central nervous system regulation of organismal energy and glucose homeostasis
Nature Metabolism (2021)
-
The Placental Transcriptome in Late Gestational Hypoxia Resulting in Murine Intrauterine Growth Restriction Parallels Increased Risk of Adult Cardiometabolic Disease
Scientific Reports (2019)
-
Neonatal growth restriction-related leptin deficiency enhances leptin-triggered sympathetic activation and central angiotensin II receptor-dependent stress-evoked hypertension
Pediatric Research (2016)
-
Maternal exposure to high-fat and high-fructose diet evokes hypoadiponectinemia and kidney injury in rat offspring
Clinical and Experimental Nephrology (2016)
