
Abstract
Ataxia with isolated vitamin E deficiency is a rare autosomal recessive neurodegenerative disease due to mutations in the α-tocopherol transfer protein gene. In ataxia with isolated vitamin E deficiency, the biochemical hallmark is the low plasmatic levels of vitamin E and, in most of the patients, vitamin E supplementation allows a stabilization of the neurologic conditions. We have investigated the genetic cause of ataxia and reduced levels of vitamin E, and apolipoproteins A1 and B in a 16-y-old patient. Results revealed that our propositus is a compound heterozygote for the c.227_229delinsATT/c.744delA mutations in the α-tocopherol transfer protein gene, each inherited from one of the two parents. His sister is also a compound heterozygote for both mutations, and she presents a biochemical pattern similar to that of his brother. After receiving the vitamin E supplementation, plasmatic levels of vitamin E and apolipoprotein A1 have been normalized in the propositus. The detected mutations would justify the undetectable levels of vitamin E, but would not explain the also decreased levels of the apolipoproteins, as neither that after treatment with vitamin E, the levels of apolipoprotein B do not become normal. These findings suggest that other genes may play a role in producing this atypical biochemical profile.
Similar content being viewed by others

Main
Ataxia with isolated vitamin E deficiency (AVED; MIM 277460) is a rare autosomal recessive neurodegenerative disease, mostly detected in the Mediterranean basin. AVED is due to mutations in the α-tocopherol transfer protein (α-TTP) gene placed on chromosome 8q (1). In North-African and South-Italian populations, the most frequent mutation responsible for the disease is the c.744delA mutation, whereas in AVED families of North European origin, the c.513insTT mutation has been often characterized (1,2). Because the α-TTP gene was first identified as the defective gene, in this disease, nearly 18 different mutations in the α-TTP gene have been reported and they are placed along the entire gene (1–8).
Clinical features include age at onset before 20 y gait and limb ataxia, dysarthria, lower limb areflexia, loss of vibration and positional sense, and bilateral extensor plantar reflexes. The deficit reduces the capacity to incorporate α-tocopherol (the most biologically active form of vitamin E) into very low-density lipoproteins secreted by the liver and therefore into plasma and tissues. In fact, in AVED, the sole and primary biochemical abnormality has very low levels of vitamin E in plasma (<3 mg/L; reference values 3–15 mg/L), in the absence of intestinal fat malabsorption and/or abetalipoproteinemia (ABL) (9). The vitamin E supplementation allowed a stabilization of the neurologic conditions in most of the patients (10), which suggests that a prompt genetic characterization of AVED may promote an early effective treatment of the disease.
METHODS
Patients.
The propositus is currently a 16-y-old male, born of unrelated parents, and without any familial history of epilepsy, mental retardation, or neurodegenerative disorders. The first symptoms were observed at the age of 3 y; he showed progressive ataxic gait. Physical examination showed that he was well nourished but hypotonic. All senses including positional and vibratory were normal. Lower limbs reflexes were weak whereas upper limbs reflexes were normal and symmetric. Romberg's test was negative, and he had a slight head and intention tremor. Brain magnetic resonance imaging (MRI) showed no signs of anomaly. Electrophysiological examination of sural and peroneal nerves showed normal action potentials and conduction velocities. Visual and trunk evoked potentials were normal, but somato-sensorial evoked potentials findings were consistent with an alteration of the sensory way in the spinal cord (data not shown). Cardiac examination and ECG were normal. Funduscopy (eye exam) was also normal. Hematological and biochemical tests were normal except for vitamins A and E, and apolipoproteins A1 (apoA1) and B (apoB) (Table 1). Patient is currently receiving treatment with high doses of vitamin E. Plasmatic levels have been normalized (Table 1) and neurologic symptoms have been stabilized. The 12-y old sister did not refer any symptom except for a light head titubation and intention tremor. MRI was normal and plasmatic vitamin E, apoA1 and apoB levels were reduced (Table 1), suggesting that she could also be affected. She is currently receiving vitamin E supplementation. The propositus and his family were aware of the investigative nature of the studies and gave their consent. Investigations were approved by the Scientific Committees of both Hospital of Mérida and Instituto de Biomedicina de Valencia.
Mutation analysis.
Genomic DNA from the patient, his sister, and his parents were obtained by standard methods. Mutation analysis of the α-TTP gene was performed by direct sequencing of each exon and their exon-intron boundaries of purified PCR products (Qiagen, Hilden, Germany) amplified using primers as described elsewhere (7) in an ABI Prism 3130xl autoanalyzer (Applied Biosystems, Foster City, CA).
RESULTS
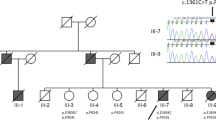
Clinical and biochemical data, mainly the extremely low levels of plasmatic vitamin E, pointed toward the diagnosis of AVED. The analysis of the α-TTP gene revealed that the patient was compound heterozygote for the mutations c.227_229delinsATT (p.W76_R77delinsYX) in exon 2 and c.744delA (p.E248fs) in exon 5 (Fig. 1). The deletion-insertion (delins) change consists of three consecutive novel heterozygous missense mutations and predicts a truncated protein. Healthy parents were heterozygous carriers: the mother carried the peculiar c.227_229delinsATT mutation and the father harbored the so frequent frameshift c.744delA mutation. Both changes were also analyzed in his sister. Results showed that she was also a compound heterozygote for the α-TTP p.W76_R77delinsYX/p.E248fs mutations.

(A) Family tree and mutation inheritance. The mutations carried by each individual have been indicated. Healthy parents carried one of the two detected mutations, c.744delA or c.227_229delinsAT, in a heterozygous state and both siblings had the two identified mutations in heterozygosis. A black arrow indicates the proband. (B) Electrophoregrams of the two identified mutations in the α-TTP gene. On the left, the electrophoregrams corresponding to a healthy control and to the proband with the c.227_229delinsAT mutation in heterozygosis. Three red arrows point at the three consecutive changes: [227G>A + 228G>T + 229A>T]. On the right, the electrophoregrams corresponding to a healthy control and to the proband with the c.744delA mutation in heterozygosis. A red arrow shows where the frameshift starts.
DISCUSSION
The α-TTP c.744delA mutation is the most common alteration in AVED patients from the Mediterranean basin, accounting for 68% of the mutant alleles (1,2). By contrast, the α-TTP c.227_229delinsATT mutation is a novel triple missense mutation that consists of three base pairs (bp) deletion, GGA, and three bp insertion, ATT, and generates a premature stop codon (p.W76_R77delinsYX). The α-TTP protein is a member of the family of lipid-binding protein containing two CRAL-TRIO domains, pfam03765 (residues 11–83) and pfam00650 (residues 89–275). The substrate of the α-TTP protein, the α-tocopherol, is bound in a hydrophobic pocket that is constructed by residues belonging to the second domain of the α-TTP. Consequently, a protein with the p.W76_R77delinsYX mutation could not be functionally active.
Many mutations of different types have been described along the α-TTP gene (The Human Gene Mutation Database, HGMD, http://www.hgmd.cf.ac.uk/ac/index.php; Fig. 2), although only one small delins mutation is known: c.530_532delAGinsGTAAGT (1). For this reason, the complex α-TTP c.227_229delinsATT mutation is itself a striking finding. The HGMD contains 57,047 mutations belonging to 2183 genes (release March 2008). The most common types of mutation causing human genetic disease are missense and nonsense mutations (57.13%), whereas the delins (1.47%) is an uncommon type of mutation (11). Delins appear to represent combined microdeletion/microinsertion events that give rise to the replacement of one or more bp by others, not necessarily the same number. Sorted complex mutations and also small delins mutations have been described in many different genes. A three bp delins mutation, c.2540_2542delinsTCC (p.Q847_K848delinsLQ), has been described in the GUCY2D (guanylate cyclase 2D) gene, which is responsible for a cone-rod dystrophy (12). The GUCY2D gene appears to be prone to complex missense mutations such as the p.E837_R838_T839delinsDCM mutation (13) and the p.E837_R838delinsDS mutation (14). It has been suggested that the hypermutability of CpG dinucleotides could be the cause of these complex mutations (14). By contrast, the GUCY2D c.2540_2542delinsTCC change is not placed in a CpG rich region, as neither the c.227_229delinsATT mutation in the α-TTP.

Distribution of the identified α-TTP gene mutations. We have identified two mutations in our proband: the novel c.227_229delinsATT (p.W76_R77delinsYX) mutation in exon 2 and the so frequent c.744delA (p.E248fs) frameshift in exon 5.
The rich CpG sequences are not the only hotspot regions with a high degree of mutability. Overrepresentation of some penta-, tetra-, and trinucleotide motifs in the vicinity of delins has been described (15). In fact, an accurate analysis of the close sequence to the α-TTP c.227_229delinsATT mutation shows that some of these motifs are present, such as the WNRGT, TARGY, and TARSY. In the α-TTP gene other delins mutation has been reported, c.530_532delinsGTAAGT (1), and also some of these motifs, TAGSY and TARSY, are present. Moreover, Krawczak and Cooper (16) identified a consensus sequence (TGRRKM), present in several micro-deletion hotspots, which are found to be strikingly similar to putative DNA polymerase α-arrest sites. This consensus sequence is also present where the α-TTP c.227_229delinsATT mutation occurs. Based on the types of repeat involved, several mechanisms of mutation have been reported to explain the delins changes. In a delins, there is an initial mutational event being followed by a partially successful attempt at restoring the original sequence and the complexity of a given fragment should remain more or less the same as a net consequence of the combined delins event (15). This point is confirmed in the α-TTP c.227_229delinsATT mutation, as the overall complexity of the sequence is not extremely modified.
Patients with mutations in the α-TTP gene have impaired the ability to incorporate α-tocopherol, the most biologic active form of vitamin E, into lipoproteins secreted by the liver (17). Because both mutations generate a premature stop, no functionally active protein is expected to be present in the patient's cells what would explain the undetectable levels of vitamin E. Interestingly, apolipoproteins levels are also decreased in our propositus and moreover, it is striking that apoA1 levels, but not apoB, becomes normal after treatment with vitamin E (Table). This is not a frequent association in AVED patients. One plausible explanation could be that other genes are involved. Because of the so low levels of apoB in both sibs, a concomitant familial hypobetalipoproteinemia (FHBL; OMIM 107730) or even, ABL (OMIM 200100) were suspected. FHBL patients have reduced levels of apoB and LDL-C and have mutations in the APOB gene. ABL patients show low levels of total cholesterol, VLDL, LDL and an almost complete absence of apoB, and present mutations in the MTP gene. We searched for mutations in both APOB and MTP genes in the propositus, but no changes were found. Maybe, other still unidentified genes linked to FHBL (18) could explain the biochemical profile in both sibs. Recently, the PCSK9 gene that cosegregates with hypercholesterolemia (OMIM 603776) has been proposed as a candidate gene in FHBL (19), although searching for mutations in the PCSK9 gene did not yield any positive result in our patient. In any case, it is tempting to speculate that other genes may play a role in the atypical biochemical profile detected in our patients.
Abbreviations
- α-TTP:
-
α-tocopherol transfer protein
- apo:
-
apolipoprotein
- AVED:
-
ataxia with isolated vitamin E deficiency
- bp:
-
base pair
- delins:
-
deletion-insertion
- FHBL:
-
familial hypobetalipoproteinemia
- GUCY2D:
-
guanylate cyclase 2D
References
Ouahchi K, Arita M, Kayden H, Hentati F, Ben Hamida M, Sokol R, Arai H, Inoue K, Mandel JL, Koenig M 1995 Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet 9: 141–145
Cavalier L, Ouahchi K, Kayden HJ, Di Donato S, Reutenauer L, Mandel JL, Koenig M 1998 Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet 62: 301–310
Gotoda T, Arita M, Arai H, Inoue K, Yokota T, Fukuo Y, Yazaki Y, Yamada N 1995 Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N Engl J Med 333: 1313–1318
Hentati A, Deng HX, Hung WY, Nayer M, Ahmed MS, He X, Tim R, Stumpf DA, Siddique T, Ahmed 1996 Human alpha-tocopherol transfer protein: gene structure and mutations in familial vitamin E deficiency. Ann Neurol 39: 295–300
Tamaru Y, Hirano M, Kusaka H, Ito H, Imai T, Ueno S 1997 Alpha-Tocopherol transfer protein gene: exon skipping of all transcripts causes ataxia. Neurology 49: 584–588
Shimohata T, Date H, Ishiguro H, Suzuki T, Takano H, Tanaka H, Tsuji S, Hirota K 1998 Ataxia with isolated vitamin E deficiency and retinitis pigmentosa. Ann Neurol 43: 273
Hoshino M, Masuda N, Ito Y, Murata M, Goto J, Sakurai M, Kanazawa I 1999 Ataxia with isolated vitamin E deficiency: a Japanese family carrying a novel mutation in the alpha-tocopherol transfer protein gene. Ann Neurol 45: 809–812
Usuki F, Maruyama K 2000 Ataxia caused by mutations in the alpha-tocopherol transfer protein gene. J Neurol Neurosurg Psychiatry 69: 254–256
Koenig M 2000 Ataxia with isolated vitamin E deficiency. Klockgether T Handbook of Ataxia Disorders. New York Marcel Dekker, Inc pp 223–234
Mariotti C, Gellera C, Rimoldi M, Mineri R, Uziel G, Zorzi G, Pareyson D, Piccolo G, Gambi D, Piacentini S, Squitieri F, Capra R, Castellotti B, Di Donato S 2004 Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci 25: 130–137
Antonarakis SE, Krawczak M, Cooper DN 2001 The nature and mechanisms of human gene mutation. Scriver CR, Beaudet AL, Sly WS, Valle D The Metabolic and Molecular Bases of Inherited Disease. New York, McGraw-Hill pp 343–377
Yoshida S, Yamaji Y, Yoshida A, Kuwahara R, Yamamoto K, Kubata T, Ishibashi T 2006 Novel triple missense mutations of GUCY2D gene in Japanese family with cone-rod dystrophy: possible use of genotyping microarray. Mol Vis 12: 1558–1564
Perrault I, Rozet JM, Gerber S, Kelsell RE, Souied E, Cabot A, Hunt DM, Munnich A, Kaplan J 1998 A retGC-1 mutation in autosomal dominant cone-rod dystrophy. Am J Hum Genet 63: 651–654
Payne AM, Morris AG, Downes SM, Johnson S, Bird AC, Moore AT, Bhattacharya SS, Hunt DM 2001 Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J Med Genet 38: 611–614
Chuzhanova NA, Anassis EJ, Ball EV, Krawczak M, Cooper DN 2003 Meta-analysis of indels causing human genetic disease: mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat 21: 28–44
Krawczak M, Cooper DN 1991 Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet 86: 425–441
Traber MG, Sokol RJ, Burton GW, Ingold KU, Papas AM, Huffaker JE, Kayden HJ 1990 Impaired ability of patients with familial isolated vitamin E deficiency to incorporate alpha-tocopherol into lipoproteins secreted by the liver. J Clin Invest 85: 397–407
Schonfeld G, Lin X, Yue P 2005 Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci 62: 1372–1378
Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D, Bocchi L, Valenti V, Bonardi R, Guardamagna O, Averna M, Tarugi P 2007 A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol 27: 677–681
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Spanish Network on Cerebellar Ataxias of the Instituto de Salud Carlos III (Grant G03/56). The CIBERER is an initiative of the Instituto de Salud Carlos III.
Rights and permissions
About this article
Cite this article
Fernández-Burriel, M., Martínez-Rubio, D., Lupo, V. et al. A Novel Delins Mutation in the α-TTP Gene in a Family Segregating Ataxia With Isolated Vitamin E Deficiency. Pediatr Res 64, 262–264 (2008). https://doi.org/10.1203/PDR.0b013e31817d9bf7
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31817d9bf7
This article is cited by
-
A novel delins (c.773_819+47delinsAA) mutation of the PCCA gene associated with neonatal-onset propionic acidemia: a case report
BMC Medical Genetics (2020)
-
Ataxia with vitamin E deficiency: update of molecular diagnosis
Neurological Sciences (2010)
