
Abstract
A 10-wk-old infant girl with severe hypertrophy of the septal and atrial walls by cardiac ultrasound, developed progressive ventricular wall thickening and died of aspiration pneumonia at 5 mo of age. Postmortem examination revealed ventricular hypertrophy and massive atrial wall thickening due to glycogen accumulation. A skeletal muscle biopsy showed increased free glycogen and decreased activity of phosphorylase b kinase (PHK). The report of a pathogenic mutation (R531Q) in the gene (PRKAG2) encoding the γ2 subunit of AMP-activated protein kinase (AMPK) in three infants with congenital hypertrophic cardiomyopathy, glycogen storage, and “pseudo PHK deficiency” prompted us to screen this gene in our patient. We found a novel (R384T) heterozygous mutation in PRKAG2, affecting an arginine residue in the N-terminal AMP-binding domain. Like R531Q, this mutation reduces the binding of AMP and ATP to the isolated nucleotide-binding domains, and prevents activation of the heterotrimer by metabolic stress in intact cells. The mutation was not found in DNA from the patient's father, the only available parent, and is likely to have arisen de novo. Our studies confirm that mutations in PRKAG2 can cause fatal infantile cardiomyopathy, often associated with apparent PHK deficiency.
Similar content being viewed by others


Main
The most common cause of cardiac glycogenosis in infancy is Pompe disease (glycogen storage disease type II, GSD II), due to deficiency of the lysosomal enzyme acid α-1,4-glucosidase (1). The only other glycogenosis causing massive and rapidly fatal cardiomegaly in infants is a variant of phosphorylase b kinase (PHK) deficiency, which has been reported only in a handful of patients (2–7). In these cases, and contrary to what happens in GSD II, glycogen does not accumulate within lysosomes but is free in the cytoplasm of cardiomyocytes, and the disease seems to be restricted to the heart. The molecular basis of this disorder has been a puzzle until recently because none of the several isozymes of PHK is heart-specific or predominantly expressed in the heart (8). The riddle was solved when, in three unrelated sporadic patients, Burwinkel et al. (9) found an identical heterozygous pathogenic mutation (R531Q) in the gene encoding the γ2-subunit of AMP-activated protein kinase (PRKAG2). This finding prompted us to revisit an infant with fatal infantile cardiac glycogenosis and PHK deficiency that we had briefly described in 1998. We found a novel heterozygous mutation in PRKAG2, thus confirming the seemingly paradoxical association between an enzyme defect and a mutation in an unrelated gene.
PATIENTS AND METHODS
Patient.
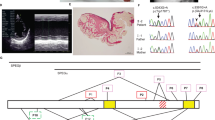
This baby girl was born at term by caesarian section (due to fetal bradycardia) to nonconsanguineous parents without any family history of cardiac disease or fetal loss. A cardiac ultrasound at 10 wk of age showed severe hypertrophy of both septal (1.52 cm) and atrial (1 cm) walls (Fig. 1, A and B). Left ventricular ejection fraction was 81%, and peak instantaneous left ventricular outflow gradient was 75 mm Hg. Electrocardiography (ECG) showed a short PR-interval, right atrial enlargement, biventricular hypertrophy and inferior-lateral ischemia (Fig. 2). Metabolic studies in cultured skin fibroblasts ruled out Pompe disease and defects of β-oxidation. A skeletal muscle biopsy showed increased glycogen not contained within lysosomes, suggesting the diagnosis of cardiac PHK deficiency. The child developed progressive ventricular wall thickening and failure to thrive, despite intensified nutritional support. She was on the waiting list for heart transplantation when she died at 5 mo of age from aspiration pneumonia. With the parent's permission, an autopsy was performed 45 min after death and frozen tissues were obtained for biochemical analyses. Again with parental consent, molecular studies were later performed in tissue from the patient and in blood from her father. This study was approved by the Institutional Review Board of Columbia University Medical Center.

2-dimensional echocardiography. (A) massively thickened intra-atrial septum. IAS, intra-atrial septum; RA, right atrium; LA, left atrium. (B) long axis view of the ventricular chambers, showing generalized right ventricular (RV) and left ventricular (LV) hypertrophy. RVFW, right ventricular free wall; PW, left ventricular posterior wall. Hatched markers are 0.5 cm apart.

Resting 12-lead ECG showing generalized ischemia, biventricular hypertrophy, and a very short PR-interval.
Morphology.
Heart and skeletal muscle samples were snap-frozen in liquid nitrogen-cooled isopenthane and cryosections were stained with a standard battery of histologic and histochemical reactions (10).
Biochemistry.
Glycogen concentration and the activities of phosphofructokinase and phosphorylase b kinase were measured in whole heart (glycogen) or in 10,000 g supernatants of 10% cardiac muscle homogenates, as described (3).
Removal of paraffin from tissue sections.
Shavings (10 μm-thick) of heart and muscle embedded in paraffin were incubated in 1 mL xylene at 55°C for 15 min. The samples were centrifuged at 12,000 g and the pellet was washed twice with 1 mL of 1/1 xylene/ethanol, twice with 1 mL absolute ethanol, then dried at 55°C for 10 min.
DNA extraction and PCR amplification.
Genomic DNA extraction was performed on de-paraffinized tissue pellets according to standard procedures (11). DNA (0.1 μg) was used for the PCR amplification of Exon 11 of the PRKAG2 gene using the following primers:
Forward, 5′-TGT AGC TTG CAC TTG TAT TTG AGA; Reverse, 5′-TGA GGG TAA CAG GAG CCA AT.
Sequence analysis of the PCR fragments.
a 417-base pair PCR fragment was separated on 2% agarose gel and purified by Perfectprep® Gel Cleanup kit (Eppendorf, Hamburg, Germany). Purified DNA fragments were sequenced with the ABI Prism 3130 Genetic Analyzer (Perkin Elmer Corp, Boston, MA).
Effect of the R384t mutation on nucleotide binding.
Plasmids encoding a glutathione-S-transferase (GST) fusion with both nucleotide-binding domains of human AMP-activated protein kinase γ2 subunit (AMPK-γ2) (cysthathionine β synthase [CBS] motifs 1-4) were expressed in bacteria, and purified on glutathione-Sepharose (12). The R384T mutation was introduced using the QuikChange site-directed mutagenesis system (Stratagene). The purified GST fusion protein was incubated with glutathione-coupled scintillation proximity assay (SPA) yttrium silicate beads (GE Healthcare), preblocked with 5% gelatin from cold-water fish skin (Sigma Chemical Co.). The beads were washed with buffer A (50 mM HEPES, pH 7.4, 200 mM NaCl and 1 mM DTT) and resuspended to 10 mg/mL. Wells in a 96 well plate were set up with varying concentrations of [3H] AMP or ATP and 0.1 mg SPA beads, and made up to a final volume of 100 μL with buffer A. After shaking the plate gently for 15 min, the beads were allowed to settle, and scintillations were counted using a Microbeta 1450 (Perkin-Elmer). Values obtained with blanks containing purified GST without the AMPK-γ2 insert were subtracted.
Effect of the R384t mutation on AMPK activity.
The R384T mutation was introduced into a pcDNA3-based vector (12) using mutagenesis as above. Co-expression in HEK-293 cells with myc-tagged α1 and β1, immunoprecipitation of recombinant complexes using anti-myc antibody, and kinase assays in the resuspended immunoprecipitate were as described previously (12).
RESULTS
Pathologic, biochemical, and molecular findings.
The heart showed marked ventricular hypertrophy (Fig. 3A), and the right atrial wall was extremely thickened, measuring over 1 cm (Fig. 3B). Staining with periodic acid Schiff (PAS) showed greatly increased glycogen content in the myocardium (Fig. 3C) and, to a lesser extent, in skeletal muscle (Fig. 3E). In both tissues, glycogen was digested normally by diastase (Fig. 3D and F). Electron microscopy confirmed the presence of large pools of normal-looking glycogen β-particles, which appeared to disrupt or distort the contractile system of the cardiomyocytes (not shown).

Gross pathology of the patient's heart, showing marked ventricular hypertrophy (A), and massive thickening of the right atrial wall (B), which exceeds 1 cm (the scale is in millimeter divisions). The PAS stain shows glycogen accumulation, which is massive in the myocardium (C) and less severe in skeletal muscle (E). Glycogen is completely digested by diastase in both heart (D) and muscle (F).
Biochemical assays in postmortem myocardium (frozen within 2 h of death) showed an 8-fold increase in glycogen concentration and undetectable PHK activity. To reassure ourselves that the lack of PHK activity was not a postmortem artifact, we measured the activity of phosphofructokinase (PFK), a notoriously labile glycolytic enzyme, which was not much less in the patient's heart than in two hearts obtained at transplantation from patients without metabolic disorders (Table 1).
Given the clinical, histochemical, and biochemical similarities between this patient and those reported by Burwinkel et al. (9), we did not sequence any of the genes encoding subunits of PHK but sequenced instead the PRKAG2 gene, encoding the γ2 subunit of AMPK. Sequence analysis revealed a single heterozygous G-to-C change at nucleotide position 1151 (Fig. 4A). This transition predicts the substitution of an arginine by a threonine at codon 384 (Fig. 4B and C).

(A) Electropherograms of part of exon 11 of the PRKAG2 gene isolated from the patient's heart. A single G-to-C change at nucleotide position 1151 (asterisk) changes an arginine (Arg) to a threonine (Thr). (B) The region of PRKAG2 containing the R384T mutation is aligned with sequences of PRKAG from other species. (C). Schematic view of the PRKAG2 protein showing the four CBS motifs and the site of the R384T mutation (red arrow). The other arrows show the sites of pathogenic mutations reported in other patients with cardiac glycogenoses.
Effect of the R384t Mutation on the Regulation of AMPK.
The γ2 mutation occurs within the second of four cystathionine β-synthase (CBS) motifs that form the regulatory AMP/ATP binding sites of AMPK (13). To study the effects of the mutation on the regulation of AMPK by these nucleotides, we first expressed both the wild-type sequence and an R384T mutation of the four CBS motifs from γ2 as glutathione-S-transferase (GST) fusions in bacteria and purified them on glutathione-Sepharose. Binding of AMP and ATP was then assessed using a new scintillation proximity assay. In the presence of the R384T mutation, the concentration of AMP needed to cause half-maximal binding (B0.5) rose from 41 ± 11 μM to 700 ± 130 μM, a 17-fold increase (Fig. 5A). The R384T mutation increased the B0.5 for ATP by an even larger amount (26-fold), from 160 ± 40 μM to 4030 ± 730 μM (Fig. 5A).

(A) Binding of AMP, measured by scintillation proximity, by the GST-γ2 fusion protein with the wild type sequence (open circles) or the R384T mutation (filled circles). Data were fitted to the equation: Binding = Bmax · [AMP]/(B0.5 + [AMP]) using GraphPad Prism, and the curves were generated using the best-fit parameters obtained. (B) As (A), but showing binding of ATP. (C). Activity, with and without 200 μM AMP, of recombinant α1β1γ2 complexes with wild type sequence and R384T mutation, expressed in HEK-293 cells and extracted under “slow lysis” and “rapid lysis” conditions.
To investigate the effect of the mutation on the regulation of AMPK activity, we also introduced the mutation in a pcDNA3-based plasmid encoding full-length human γ2 (long form) and co-expressed it with myc-tagged α1 and β1 in human embryonic kidney (HEK-293) cells. AMPK activity was determined in recombinant AMPK complexes immunoprecipitated with anti-myc antibodies (which do not precipitate endogenous AMPK). We harvested the cells both by the “rapid lysis” procedure, which preserves the physiologic phosphorylation state of the regulatory site Thr-172, and by the “slow lysis” procedure, which (at least with the wild type enzyme) yields maximal phosphorylation of Thr-172 due to stresses that occur during cell harvesting (12). After “slow lysis”, we measured activity with and without 200 μM AMP to see if the mutant could be allosterically activated by the nucleotide. Figure 5C shows that, as expected, slow lysis caused a 10-fold activation (in the presence of AMP) of the wild-type enzyme compared with rapid lysis (p < 0.001), and the enzyme harvested by slow lysis was also 70% stimulated by 200 μM AMP (p < 0.001). As shown in Fig. 5, the basal activity of the R384T mutant after rapid lysis was not significantly different from that of the wild-type, and neither slow lysis nor addition of AMP in the assay caused significant increases in activity.
DISCUSSION
Our patient had prenatal onset (fetal bradycardia leading to caesarian section) of a severe and rapidly fatal infantile hypertrophic nonobstructive cardiomyopathy with massive cardiomegaly due to glycogen storage. The most common cause of neonatal cardiac glycogenosis, Pompe disease, was unlikely on clinical grounds (infants with Pompe disease are floppy, have hepatomegaly and often macroglossia) and was excluded by the normal α-glucosidase activity in fibroblasts. A second, less frequent cause of congenital cardiac glycogenosis has been ascribed to deficiency of PHK, a cytoplasmic enzyme involved in the activation of phosphorylase (2–4,7). The similarities between the clinical and pathologic features of this patient and those of another infant with PHK deficiency (3) prompted us to measure PHK activity in frozen postmortem myocardium and led us to diagnose PHK-deficient cardiomyopathy and to postulate the defect of a gene encoding a PHK isozyme expressed predominantly, if not exclusively, in the heart (Bruno C et al. 3rd International Congress of the World Muscle Society, Naples, May 29–30, 1998).
PHK deficiency has been associated with five main syndromes distinguished by inheritance and by tissue involvement: (i) a benign X-linked recessive liver disease of infancy or childhood (14); (ii) an autosomal recessive liver and muscle disease (15); (iii) a pure myopathy affecting both sexes but predominantly men (16); (iv) an autosomal recessive severe liver disease associated with cirrhosis (17) and (v) the fetal infantile cardiopathy described here.
PHK is a multimeric enzyme composed of four different subunits, α, β, γ, and δ. The enzyme composition is (αβγδ)4. The γ subunit is catalytic and is regulated through the degree of phosphorylation of the α and β subunits. Calcium sensitivity is conferred by the δ subunit, which is a tightly bound calmodulin. The involvement of different tissues in the various clinical variants of PHK deficiency is explained by the multiplicity of genes encoding the various subunits and their isoforms, with two genes encoding the α subunit, one the β subunit, two the γ subunit, and three encoding calmodulin (14). The benign X-linked hepatopathy is due to mutations in PHKA2, which encodes a nonmuscle α isozyme. The autosomal recessive liver and muscle form of PHK deficiency is due to mutations in the β subunit, which is encoded by a gene on chromosome 16 (15). At least some patients with myopathy have mutations in the PHKA1 gene, which is on the proximal long arm of the × chromosome and encodes the muscle α isozyme (18,19).
The genetic basis of the “cardiopathic” PHK deficiency has always presented a conundrum because there is no heart-specific PHK isozyme. This riddle was solved when Burwinkel et al. (9) definitively excluded mutations in any of the PHK genes and instead identified, in three of five unrelated patients with the disease, a single mutation in the gene (PRAKG2) encoding the γ2 subunit of the AMP-activated protein kinase (AMPK) (9). Thus, the blame was shifted from one complex kinase, PHK to another, AMPK.
AMPK is an αβγ heterotrimer, where α is the catalytic subunit and β and γ are regulatory subunits (20–22). It is considered a “cellular fuel gauge”, which is switched on by increases in the AMP:ATP ratio, an indicator of cellular energy deficit. Once activated, AMPK switches off ATP-consuming anabolic pathways (e.g., glucose, lipid, and protein synthesis) while switching on ATP-generating catabolic pathways (e.g., uptake and oxidation of glucose and fatty acids). Increases in AMP activate the AMPK complex via two mechanisms. Firstly, AMPK is constitutively phosphorylated at a specific threonine residue (Thr-172) within the α subunit by the tumor suppressor protein kinase LKB1 (23,24); this phosphorylation is required for activity (25,26), but dephosphorylation of Thr-172 is inhibited by AMP (12,27). Secondly, AMP further activates the phosphorylated enzyme via an allosteric mechanism (28).
The γ subunits of AMPK contain four tandem repeats of a sequence named by Bateman a CBS motif (29) after the enzyme cystathionine β-synthase, in which two such repeats occur. CBS motifs act in pairs to form domains now called “Bateman domains” that bind adenosine-containing ligands (13). The four CBS motifs in the γ subunits of AMPK form two domains, each of which binds one molecule of the regulatory ligand, AMP or ATP, in a mutually exclusive manner (13).
Several groups (30–34) reported pathogenic mutations in PRKAG2 in adult patients with autosomal dominant hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome (FHC/WPW), which was considered “a novel glycogen storage of the heart” (30). The heterozygous mutation found by Burwinkel et al. (9) in the three sporadic cases of infantile cardiomyopathy, R531Q, was not found in any adult patients, although a different mutation affecting the same residue (R531G) was identified in a family with FHC/WPW and childhood onset (31). The R531Q and R531G mutations neutralize the positively charged side chain of an arginine residue that occurs in the C-terminal Bateman domain, and we have previously reported that these mutations reduce the binding of AMP and ATP (9,14). Intriguingly, the mutation in our patient (R384T) neutralizes the positively charged side chain of an arginine residue in the N-terminal Bateman domain that exactly aligns with Arg-531 in the C-terminal Bateman domain. R384 is also adjacent to H383, in which mutation to arginine has been reported to cause adult onset WPW/HCM (32). Modeling of the Bateman domains (based on the crystal structure of a bacterial enzyme containing similar domains) suggests that these positively charged, basic side chains (H383, R384 and R531) are directly involved in the binding of the negatively charged α-phosphate groups of AMP or ATP (14). Consistent with this, the H383R, R531G and R531Q mutations all interfere with the binding of AMP and ATP to the isolated Bateman domains, and prevent activation of recombinant αβγ complexes by AMP (9,14).We now show that the R384T mutation has similar effects.
In previous studies, the R531G and R531Q mutations not only prevented activation by AMP but also caused an increase in the basal activity of the αβγ complex, because, we proposed, the mutations interfered with the binding of the inhibitory nucleotide, ATP (9). Although the R384T mutation also caused a marked reduction in ATP binding (Fig. 5B), we did not observe any increase in the basal activity of the α1β1γ2 complex expressed in HEK293 cells (Fig. 5C).
The three cases of R531Q mutation reported by Burwinkel et al. (9) were sporadic and in the one case where they were available, both parental DNAs were normal. Our patient was also a sporadic case and her father did not harbor the mutation. Although the mother could not be studied, it is likely that this was also a de novo mutation.
Our findings confirm the causative role of PRKAG2 mutations in the fatal infantile hypertrophic cardiomyopathy hitherto erroneously attributed to PHK deficiency. They also add a new “severe” mutation to that described by Burwinkel and coworkers (9). However, several questions remain unanswered, as noted by those authors (9).
-
(1) Our discovery does not completely resolve the question of the “pseudo-PHK deficient cardiopathy” of infancy, because two children in the series of five described by Burwinkel et al. (9) did not harbor mutations in PRKAG2.
-
(2) It is not clear whether AMPK deficiency is confined to the heart or also affects other tissues. By Northern blotting of human tissues, the γ2 isoform is widely expressed, although most abundant in skeletal and cardiac muscle (35). The finding of increased glycogen in the skeletal muscle biopsy from our patient and from one of the patients harboring the R531Q mutation (9) suggests a more widespread defect, although the heart is selectively affected.
-
(3) The cause of glycogen accumulation remains unclear, although Burwinkel et al. suggested that (despite the loss of AMP activation caused by the mutation) the basal phosphorylation and activity of AMPK are increased by the R531Q mutation (9). This may result in increased glucose uptake into cardiomyocytes, increased levels of glucose-6-phosphate (G6P), and activation of glycogen synthase. This scenario would also explain the presence of abnormal glycogen (polyglucosan) observed in the heart of adult patients (30), which could be due to a skewed ratio between glycogen synthase and glycogen branching enzyme, as seen in branching enzyme deficiency and also in PFK deficiency (36).
-
(4) The reasons for the apparent lack of PHK activity in tissue extracts from these patients remains unclear. AMPK was reported to phosphorylate PHK in cell-free assays (37), although the significance of this finding has been questioned (38). One trivial reason for the lack of PHK activity may be the extreme lability of the enzyme, which often has to be measured in postmortem tissues.
Irrespective of the exact pathogenesis, mutations in the γ2 subunit of AMPK have to be considered an important cause of congenital hypertrophic nonobstructive cardiomyopathy and patients should be screened for PRKAG2 mutations. Given the predominant, if not exclusive, involvement of the heart, cardiac transplantation is an option to be considered in this otherwise rapidly fatal condition.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CBS:
-
cysthathionine β synthase
- PHK:
-
phosphorylase b kinase
- PRKAG2:
-
AMP-activated protein kinase γ2 subunit
References
Engel AG, Hirschhorn R, Huie M 2004 Acid maltase deficiency. In: Engel AG, Franzini-Armstrong C (eds) Myology. McGraw Hill, New York, pp 1559–1586
Mizuta K, Hashimoto E, Tsutou A, Eishi Y, Takemura T, Narisawa K, Yamamura H 1984 A new type of glycogen storage disease caused by deficiency of cardiac phosphorylase kinase. Biochem Biophys Res Commun 119: 582–587
Servidei S, Metlay LA, Chodosh J, DiMauro S 1988 Fatal infantile cardiopathy caused by phosphorylase b kinase deficiency. J Pediatr 113: 82–85
Elleder M, Shin YS, Zuntova A, Vojtovic P, Chalupecky V 1993 Fatal infantile hypertrophic cardiomyopathy secondary to deficiency of heart specific phosphorylase b kinase. Virchows Arch A Pathol Anat Histopathol 423: 303–307
Regalado JJ, Rodriguez MM, Ferrer PL 1999 Infantile hypertrophic cardiomyopathy of glycogenosis type IX: isolated cardiac phosphorylase kinase deficiency. Pediatr Cardiol 20: 304–307
Buhrer C, van Landeghem FK, Felderhoff-Mueser U, Stadelmann C, Obladem M 2003 Fetal bradycardia at 28 weeks of gestation associated with cardiac glycogen phosphorylase b kinase deficiency. Acta Paediatr 92: 1352–1353
Eishi Y, Takemura T, Sone R, Yamamura H, Narisawa K, Ichinohasama N, Tanaka M, Hatakeyama S 1985 Glycogen storage disease confined to the heart with deficient activity of cardiac phosphorylase kinase: a new type of glycogen storage disease. Hum Pathol 16: 193–197
Burwinkel B, Hu B, Schroers A, Clemens PR, Moses SW, Shin YS, Pongratz D, Vorgerd M, Kilimann MW 2003 Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet 11: 516–526
Burwinkel B, Scott JW, Buhrer C, van Landeghem FK, Cox GF, Wilson CM, Hardie DG, Kilimann MW 2005 Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 76: 1034–1049
Dubowitz V 1985 Muscle biopsy. A practical approach. Bailliere Tindall, Philadelphia, pp 29–38
Sambrook J, Russell DW 2001 Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 6.4–6.11
Davies SP, Helps NR, Cohen PT, Hardie DG 1995 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2Calpha and native bovine protein phosphatase-2Ac. FEBS Lett 377: 421–425
Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG 2004 CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 113: 274–284
Hendrickx J, Lee P, Keating J, Carton D, Sardharwalla IB, Tuchman M, Baussan C, Willems PJ 1999 Complete genomic structure and mutational spectrum of PHKA2 in patients with X-linked liver glycogenosis type I and II. Am J Hum Genet 64: 1541–1549
Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS, Strachan JA, Kiliman MW 1997 Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum Mol Genet 6: 1109–1115
Wilkinson DA, Tonin P, Shanske S, Lombes A, Carlson GM, DiMauro S 1994 Clinical and biochemical features of 10 adult patients with muscle phosphorylase kinase deficiency. Neurology 44: 461–466
Kilimann MW 1997 Glycogen storage disease due to phosphorylase kinase deficiency. In: Swallow DM, Edwards YH (eds) Protein Dysfunction in Human Genetic Disease. Bios Scientific Publishers, Oxford, pp 57–75
Bruno C, Manfredi G, Andreu AL, Shanske S, Krishna S, Ilse WK, DiMauro S 1998 A splice junction mutation in the alpha-M gene of phosphorylase kinase in a patient with myopathy. Biochem Biophys Res Commun 249: 648–651
Wehner M, Clemens PR, Engel AG, Kilimann MW 1994 Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet 3: 1983–1987
Hardie DG, Carling D 1997 The AMP-activated protein kinase: fuel gauge of the mammalian cell. Eur J Biochem 246: 259–273
Hardie DG 2004 The AMP-activated protein kinase pathway - new players upstream and downstream. J Cell Sci 117: 5479–5487
Hardie DG, Hawley SA, Scott JW 2006 AMP-activated protein kinase - development of the energy sensor concept. J Physiol 574: 7–15
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG 2003 Complex between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2: 28
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Sclattner U, Wallimann T, Carlson M, Carling D 2003 LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–2008
Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG 1996 Characterization of the AMP-activated protein kinase kinase from rat liver, and identification of theonine 172 as the major site at which it phosphorylates and activates AMP-activated protein kinase. J Biol Chem 271: 27879–27887
Stein SC, Woods A, Jones NA, Davison MD, Carling D 2000 The regulation of AMP-activated protein kinase by phosphorylation. Biochem J 345: 437–443
Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D 2007 Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403: 139–148
Scott JW, Ross FA, Liu JK, Hardie DG 2007 Regulation of AMP-activated protein kinase by a pseudosubstrate sequence on the gamma subunit. EMBO J 26: 806–815
Bateman A 1997 The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 22: 12–13
Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE 2002 Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 109: 357–362
Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R 2001 Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 104: 3030–3033
Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Roberts R 2001 Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 344: 1823–1831
Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H 2001 Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10: 1215–1220
Bayrak F, Komurcu-Bayrak E, Mutlu B, Kahveci G, Basaran Y, Erginel-Unaltuna N 2006 Ventricular pre-excitation and cardiac hypertrophy mimicking hypertrophic cardiomyopathy in a Turkish family with a novel PRKAG2 mutation. Eur J Heart Fail 8: 712–715
Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D 2000 Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346: 659–669
DiMauro S, Hays AP, Tsujino S 2004 Nonlysosomal glycogenoses. In: Engel AG, franzini-Armstrong C (eds) Myology. McGraw-Hill, New York, pp 1535–1558
Carling D, Hardie DG 1989 The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta 1012: 81–86
Beyer A, Kitzerow A, Crute B, Kemp BE, Witters LA, Heilmeyer LM 2000 Muscle phosphorylase kinase is not a substrate of AMP-activated protein kinase. Biol Chem 381: 457–461
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by a grant from the Muscular Dystrophy Association (S.D.M.)., J.S., J.W.S. and D.G.H. were supported by the EXGENESIS Integrated Project (LSHM-CT-2004-005272) from the European Commission, and F.A.R. by a CASE studentship funded jointly by the BBSRC (UK) and AstraZeneca.
Rights and permissions
About this article
Cite this article
Akman, H., Sampayo, J., Ross, F. et al. Fatal Infantile Cardiac Glycogenosis with Phosphorylase Kinase Deficiency and a Mutation in the γ2-Subunit of AMP-Activated Protein Kinase. Pediatr Res 62, 499–504 (2007). https://doi.org/10.1203/PDR.0b013e3181462b86
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181462b86
This article is cited by
-
A novel PRKAG2 mutation in a Chinese family with cardiac hypertrophy and ventricular pre-excitation
Scientific Reports (2017)
-
PRKAG2 mutations presenting in infancy
Journal of Inherited Metabolic Disease (2017)
-
Targeting the energy guardian AMPK: another avenue for treating cardiomyopathy?
Cellular and Molecular Life Sciences (2017)
-
AMP Deaminase 1 Gene Polymorphism and Heart Disease—A Genetic Association That Highlights New Treatment
Cardiovascular Drugs and Therapy (2014)
-
Neuromuscular Disorders of Glycogen Metabolism
Current Neurology and Neuroscience Reports (2013)
