
Abstract
Breast milk glutamate is a potential gluconeogenic substrate. However, in piglets, most dietary glutamate undergoes first pass extraction by the gut, limiting its contribution to glucose formation. The objectives of the study were to determine in preterm infants, whether dietary glutamate increases plasma [glutamate] in a dose-dependent fashion and whether glutamate carbon appears in plasma glucose to an appreciable extent. Five enterally fed infants (31 ± 0 wk; 1555 ± 131 g) (mean ± SE) were studied twice (postnatal age 10 ± 1 d and 17 ± 1 d, respectively), while receiving an intragastric infusion of glutamate (labeled to 4% by [U-13C] glutamate) at 2.4 (study 1) and 4.8 μmol/kg/min (study 2) for 1.5 h (n = 2) or 5 h (n = 3). Plasma [glutamate] was 82 ± 8 μM at baseline, and 84 ± 11 and 90 ± 13 μM after glutamate supplementation at 2.4 and 4.8 μmol/kg/min, respectively, values not different from baseline. Plasma [glutamate] was not affected by the duration of the glutamate infusion (1.5 versus 5 h). Plasma 13C glucose enrichment was only 0.3% (after 5 h ingestion of glutamate labeled to 4%) indicating insignificant contribution of dietary glutamate carbon to glucose. Thus, in premature infants, splanchnic extraction is the major fate of dietary glutamate, which is not a significant gluconeogenic substrate in these infants.
Similar content being viewed by others

Main
Glutamate is an important nonessential amino acid. It has a key role in transdeamination of amino acids; glutamine, proline, histidine, arginine, and ornithine are disposed of via conversion to glutamate (1); glutamate is a component of the important antioxidant glutathione (2) and also of polyglutamated folic acid, an essential co-factor for a number of enzymatic processes (2). Further, glutamate is an important gluconeogenic substrate that enters the citric acid cycle via α-ketoglutarate (1,3). Glutamate also is an abundant dietary amino acid. It is, for example, the most abundant amino acid in breast milk, 2 g/L (free + protein bound). Thus, a 5-kg infant consuming about 850 mL of breast milk/d will have a total glutamate intake of 350 mg/kg/d (4,5).
The potential toxicity of exogenous glutamate has been extensively discussed as a result of Olney's report (6) in the early 1970s that large bolus doses of glutamate, aspartate, and cysteine caused acute neuronal degeneration in the retina and certain periventricular nuclei outside the blood brain barrier in neonatal mice. However, despite a very large number of studies addressing dietary effects of glutamate in human children and adults, no serious adverse effects have been observed (7) and the U.S. Food and Drug Administration has classified the sodium salt of glutamate among food ingredients that are “generally recognized as safe.”
Reeds et al. (8) demonstrated that, in piglets, the major part of dietary glutamate was extracted in the splanchnic bed, mainly by the gut, whereas the appearance of glutamate in the systemic circulation was minimal. However, it is not known whether this is the case also in human newborns, particularly those born prematurely.
We have previously reported that parenterally fed newborn preterm infants can use endogenous and exogenous substrates for production of glucose via the gluconeogenic pathway during their first days of life (9–11). Because glutamate is a potentially important gluconeogenic substrate, the aim of the present study was to investigate whether intragastrically administered glutamate would provide carbon for glucose production to a significant extent. However, based on the studies by Reeds et al. (8) in piglets, we hypothesized that 1) intragastrically administered glutamate will not increase plasma glutamate concentrations in a dose-dependent fashion because of first pass metabolism in the gut, and 2) will not support gluconeogenesis in a dose dependent fashion for the same reason.
METHODS AND MATERIALS
Subjects.
The study was approved by the Institutional Review Board for Human Research at Baylor College of Medicine and the Advisory Board of the General Clinical Research Center at Texas Children's Hospital. The studies were performed following written consent by at least one parent.
Five premature infants (birth weight, 1555 ± 131 g; gestational age at birth, 31 ± 0 wk) were studied on two occasions at a postnatal age of 10 ± 1 and 17 ± 3 d, respectively. Their actual weight on the first study occasion was 1528 ± 147 g and on the second, 1722 ± 227 g. The characteristics of the infants are depicted in Table 1. At the time of the studies, all infants were tolerating enteral feedings given at 3-h intervals via a gastric tube already in place for clinical care purposes.
Study design.
Feeding breast milk or formula (see Table 2), as ordered by the attending physician, i.e. at 3-h intervals, was continued during the studies. As demonstrated by Figure 1, on the first study occasion, the infants received glutamate labeled to 4% with [U-13C]glutamate via the gastric tube (placed for clinical care purposes) at a rate of 2.4 μmol/kg/min (0.35 mg/kg/min) and on the second at 4.8 μmol/kg/min (0.70 mg/kg/min). Since the total glutamate concentration (free + protein bound) in breast milk is 2 g/L, i.e. 2 mg/mL (4,5), these infusion rates would correspond to breast milk given at 250 mL/kg/d (250 × 2 = 500 mg/d = 0.35 mg/kg/min) and 500 mL/kg/d (500 × 2 = 1000 mg/kg/d = 0.7 mg/kg/min), respectively. The intragastric infusions were started directly at completion of the last meal preceding the study and were continued for 1.5 h in two subjects and for 5 h in three subjects. Table 2 displays the total intake of glutamate via feedings and study glutamate solution, respectively.

Study design.
The unlabeled glutamate was purchased from Sigma Chemical Co.-Aldrich (Milwaukee, WI) and the [U-13C]glutamate from Cambridge Isotopes (Andover, MA). Unlabeled and labeled glutamate were mixed (to an enrichment of 4% of [U-13C]glutamate) and dissolved in sterile water by the Investigation Pharmacy at Texas Children's Hospital (30 mg/mL; 178 mosmol/L; neutral pH).
Blood samples (0.5 mL each) were obtained via heel stick just before the feeding preceding start of the study and at the end of the 1.5- and 5-h infusions of the glutamate solution, respectively. In the subjects studied for 5 h, a feeding was administered 3 h into the study. The infusion of glutamate was continued during this meal.
Analyses.
Whole blood glucose concentrations were measured immediately using an YSI glucose analyzer (YSI 2300 Stat Plus, YSI Inc., Yellow Springs, OH). Plasma was then separated and kept frozen at –80° until further analyses. For the amino acid analyses, 100 μL of plasma was mixed with an equal volume of an aqueous solution of methionine sulfone (0.4 mM in 0.1 M HCl) as internal standard and deproteinized by centrifugation at 10,000 × g for 30 min at 4° through a 3-kDa cutoff filter. The filtrate was dried and then derivatized with phenylisothiocyanate, after which the amino acids were separated using the Alliance System (Waters 2695 Separation Module and Waters 2487 dual wavelength UV absorbance detector; Waters, Milford, MA) and a Pico Tag column (3.9 × 300 mm, C-18; Waters, Milford, MA). The second study (the higher dose of glutamate: 4.8 μmol/kg/min) was not performed until the glutamate concentrations from the first study (2.4 μmol/kg/min) were available.
For analysis using gas chromatography–combustion –isotope ratio mass spectrometry (GC-C-IRMS), the plasma samples must undergo a chemical process (derivatization) to make them volatile when injected in the GC. We used the well-established glucose pentaacetate derivative to measure the enrichment of [13C]glucose (12–14) using GC-C-IRMS (Finnigan Delta Plus XL, Finnigan, Bremen, Germany) (15,14). Briefly, the sample is deproteinized with acetone, the supernatant is brought to complete dryness under nitrogen, and a mixture of acetic anhydride and pyridine in equal amounts is added (12–14). To determine the reproducibility of the GC-C-IRMS technique using the above instrument, we analyzed three standards at an enrichment level of 0.3% (i.e. the level of the enrichments found in the current study) in triplicate. The mean ± SD was 0.2969 ± 0.0012%, i.e. CV of 0.3%.
RESULTS
Plasma glutamate concentrations.
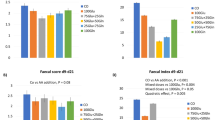
The glutamate concentrations for each individual infant are depicted in Figure 2. The graph demonstrates that glutamate concentrations vary within a wide range without relationship to either dose or duration of infusion. This was confirmed by ANOVA followed by Fisher's test (p = 0.85), which also demonstrated that the concentrations obtained after intragastric infusion at either rate were not different from baseline. Further, baseline concentrations obtained on the two study occasions were not significantly different (p = 0.6). Therefore, all baseline data (n = 10, i.e. two baselines for each subject) were averaged, 82 ± 8 μM (median, 80; range, 59–113 μM). Further, the data from all five infants (two studied for 1.5 h and three for 5 h) were grouped together. Thus, glutamate concentrations obtained after intragastric infusion of glutamate at 2.4 μmol/kg/min averaged 84 ± 11 μM (median, 76; range, 57–121 μM) and after 4.8 μmol/kg/min, 90 ± 13 μM (median, 82; range, 64–137 μM).

Individual glutamate concentrations obtained at baseline (shaded bars) and after infusion of glutamate at 2.4 (solid bars) and 4.8 (open bars) μmol/kg/min for 1.5 or 5 h.
Glucose concentrations were not affected by the intragastric glutamate infusion: 83 ± 16 (baseline study 1); 79 ± 13 (post glutamate infusion of 2.4 μmol/kg/min); 65 ± 9 (baseline study 2) and 72 ± 11 mg/dL (post glutamate infusion of 4.8 μmol/kg/min) (ANOVA followed by Fisher's test, p = 0.14).
Glucose 13C enrichment.
Since GC-C-IRMS combusts glucose carbons to CO2, our measurement of 13C content in glucose, as measured by 13CO2 enrichment above natural carbon-13 abundance, reflects the total enrichment of 13C in glucose (product) derived from the total 13C enrichment in glutamate (precursor). Table 3 depicts the 13C enrichments in glucose demonstrating that only minor increments above baseline (0.30 ± 0.04%) of the 13C enrichment in glucose were observed following the 5-h intragastric infusion of glutamate labeled with [U-13C]glutamate at the highest infusion rate, 4.8 μmol/kg/min. A negligible increase (0.09 ± 0%) was observed following 5-h infusion at the lower rate, 2.4 μmol/kg/min and no increase at all after the 1.5-h infusion at either rate.
The infants had no central or peripheral lines. Thus, the blood samples were obtained by heel stick, i.e. only small blood sample volumes could be obtained precluding analyses of glutamate enrichments.
Effects of intragastric glutamate infusion on concentrations of other amino acids.
We observed no significant changes compared with baseline in the concentrations of any of the measured amino acids (Table 4).
DISCUSSION
Our results demonstrate that intragastric supplementation of glutamate at either 2.4 or 4.8 μmol/kg/min for 1.5 or 5 h did not increase plasma glutamate concentrations above baseline, indicating that splanchnic extraction is the major fate of dietary glutamate in infants born prematurely. Thus, our results are in agreement with those reported by Reeds et al. (8) in piglets. In addition, dietary glutamate does not appear to be a significant gluconeogenic substrate in these infants.
Since the infants were fed at 3-h intervals, baseline concentrations represented 3 h post feeding conditions. At this time, the infants had no residuals, i.e. the gastric emptying was complete. In two infants, the intragastric glutamate infusion was given for 1.5 h following a feeding to determine whether the glutamate concentration would reach a peak at this time, yet this did not occur. There was no difference between baseline and values obtained after either 2.4 or 4.8 μmol/kg/min for 1.5 h. Therefore, the infusion period was extended to 5 h. However, prolonging the glutamate infusion did not affect plasma glutamate concentrations. Although our sample size is small and extrapolation to extremely low birth weight infants (birth weight <1000 g) must be done with caution, our data support the results of studies performed in children and adults (7,16) demonstrating that dietary glutamate is safe when consumed in amounts that fall within the intake ranges likely to occur with usual food intakes. Since the studies in pigs (8) show that dietary glutamate is primarily extracted by the splanchnic bed and used by the gut for fuel and as a precursor of other amino acids and metabolic products, these are also the most likely fate of glutamate in the preterm human infant. Thus, one might speculate that dietary glutamate is a potentially important fuel for the gut in premature infants, a population with high risk for intestinal problems.
The infants had no central or peripheral lines and the blood samples were, therefore, drawn by heel stick, i.e. only small amounts of blood could be obtained, permitting analyzes of only the primary outcome variables, glutamate concentrations, and glucose enrichments. However, the [U-13C]glutamate enrichment in the infusate was 4%. If one takes the unlabeled glutamate provided by the feeding immediately preceding start of the study and the feeding given 3 h into the study as well as that provided by the study infusate into account (Table 4), the precursor enrichment of [U-13C]glutamate would be diluted to 2.5% regardless of the degree of splanchnic extraction, since one has to assume that there is no difference in the extraction of labeled and unlabeled glutamate. However, taking into account that each [U-13C]glutamate molecule contains five carbons labeled with 13C, a 2.5% enrichment of [U-13C]glutamate will correspond to a 5 × 2.5% = 12.5% enrichment of carbon 13. Because GC-C-IRMS combusts glucose to CO2, our measure of 13C in glucose is represented by 13CO2 and reflects the total enrichment of 13C in glucose (product) derived from the total 13C enrichment in glutamate (precursor) = 12.5%. Thus, 0.3/12.5% = 2.4% of glutamate escaping splanchnic extraction and entering the systemic circulation would be converted to glucose. Even if all ingested glutamate were entering the systemic circulation (8 μmol/kg/min, see Table 4), the conversion of glutamate to glucose would be only 0.2 μmol/kg/min. Because the infants had no intravenous lines in place for clinical purposes at the time of the studies, for ethical reasons we did not measure glucose production (i.e. glucose derived from gluconeogenesis + glycogenolysis) using an intravenous tracer. Thus, we did not quantify the absolute contribution of glutamate carbon to glucose carbon. However assuming that glucose production is suppressed by 80% as a result of feeding (17,18) (i.e. a residual glucose production rate of 0.2 × 33 = 6.6 μmol/kg/min), the contribution from glutamate would still account for only 3% of glucose production (gluconeogenesis + glycogenolysis). Total gluconeogenesis could not be measured using [U-13C]glucose (11), as this would have required intravenous lines or deuterated water (19) and this method requires a minimum of 0.5 mL of plasma for this analysis only. Thus, we could not calculate the proportion of total gluconeogenesis derived from glutamate. However, as pointed out above, the absolute maximal possible amount of glutamate that could be converted to glucose (assuming that all ingested glutamate entered the systemic circulation) would be only 0.2 μmol/kg/min. Thus, the magnitude of the absolute gluconeogenesis from glutamate is miniscule.
The purpose of this study was to determine the role of intragastrically ingested glutamate as a gluconeogenic substrate in fed infants, i.e. under normal clinical conditions. Glutamate might play a more significant role under conditions of fasting or low energy intake. This was, however, not within the scope of this study and would have required a different study design.
In summary, the results from the present study demonstrate that intragastric supplementation of glutamate at either 2.4 or 4.8 μmol/kg/min did not increase plasma glutamate concentration above baseline. Further, intragastric glutamate carbons did not contribute to circulating glucose carbons to an appreciable extent. Thus, we conclude that splanchnic extraction is the major fate of dietary glutamate and that dietary glutamate is not a significant gluconeogenic substrate in preterm infants.
References
Brosnan JT 2000 Glutamate, at the interface between amino acid and carbohydrate metabolism. J Nutr 130: 988S–990S
Neu J 2001 Glutamine in the fetus and critically ill low birth weight neonate: metabolism and mechanism of action. J Nutr 131: 2585S–2589S
Yang D, Brunengraber H 2000 Glutamate, a window on liver intermediary metabolism. J Nutr 130: 991S–994S
Meldrum B 1993 Amino acids as dietary excitotoxins: a contribution to understanding neurodegenerative disorders. Brain Res Brain Res Rev 18: 293–314
Agostoni C, Carratù B, Boniglia C, Lammardo A-M, Riva E, Sanzini E 2000 Free glutamine and glutamic acid increase in human milk through a three months lactation period. J Pediatr Gastroenterol Nutr 31: 508–512
Olney JW, Ho O 1970 Brain damage in infant mice following oral intake of glutamate, aspartate or cysteine. Nature 227: 609–611
Walker R, Lupien JR 2000 The safety evaluation of monosodium glutamate. J Nutr 130: 1049S–1052S
Reeds PJ, Burrin DG, Stoll B, Jahoor F 2000 Intestinal glutamate metabolism. J Nutr 130: 978S–982S
Sunehag AL, Haymond MW, Schanler RJ, Reeds PJ, Bier DM 1999 Gluconeogenesis in very low birth weight infants receiving total parenteral nutrition. Diabetes 48: 791–800
Sunehag AL 2003 Parenteral glycerol enhances gluconeogenesis in very premature infants. Pediatr Res 53: 635–641
Sunehag AL 2003 The role of parenteral lipids in supporting gluconeogenesis in very premature infants. Pediatr Res 54: 480–486
Fjeld CR, Cole FS, Bier DM 1992 Energy expenditure, lipolysis, and glucose production in preterm infants treated with theophylline. Pediatr Res 32: 693–698
Sunehag A, Gustafsson J, Ewald U 1994 Very immature infants (<30 weeks) respond to glucose infusion with incomplete suppression of glucose production. Pediatr Res 36: 550–555
Sunehag AL, Treuth MS, Toffolo G, Butte NF, Cobelli C, Bier DM, Haymond MW 2001 Glucose production, gluconeogenesis and insulin sensitivity in children and adolescents: An evaluation of their reproducibility. Pediatr Res 50: 115–123
Wolfe RR 1992 Gas chromatography mass spectrometry: instrumentation. In: Wolfe RR (ed) Radioactive and Stable Isotope Tracers in Biomedicine. Principles and Practice of Kinetic Analysis. Wiley-Liss Inc, New York, pp 45–47
Geha RS, Beiser A, Ren C, Patterson R, Greenberger PA, Grammer LC, Ditto AM, Harris KE, Shaughnessy MA, Yarnold PR, Corren J, Saxon A 2000 Review of alleged reaction to monosodium glutamate and outcome of a multicenter double-blind placebo controlled study. J Nutr 130: 1058S–1062S
Sunehag A, Ewald U, Larsson A, Gustafsson J 1993 Glucose production rate in extremely immature neonates (<28 w) studied by use of deuterated glucose. Pediatr Res 33: 97–100
Bier DM, Leake RD, Haymond MW, Arnold KJ, Gruenke LD, Sperling MA, Kipnis DM 1977 Measurement of “true” glucose production rates in infancy and childhood with 6,6 - dideuteroglucose. Diabetes 26: 1016–1023
Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC 1996 Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest 98: 378–385
Acknowledgements
The authors thank our research nurses Cindy Bryant, Pamela Gordon, and Geneva Shores for excellent assistance with the studies, the Baylor General Clinical Research Center (RR00188) for its support, and Xiaoyan Chang and Shaji Chacko for invaluable technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is a publication of the U.S. Department of Agriculture/Agricultural Research Service, Children's Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX. The contents of this publication do not necessarily reflect the views or policies of the U.S. Department of Agriculture, nor does mention of trade names, commercial products, or organizations imply endorsement from the U.S. government.
Supported by the International Glutamate Technical Committee, U.S. Department of Agriculture Cooperative Agreement 58-6250-6-001, and National Institutes of Health grant MO1-RR-00188. Dr Haÿs research fellowship was partially supported by grants from Blédina and from Société Francophone de Nutrition Entérale et Parentérale.
Rights and permissions
About this article
Cite this article
Haÿs, S., Ordonez, J., Burrin, D. et al. Dietary Glutamate Is Almost Entirely Removed in Its First Pass Through the Splanchnic Bed in Premature Infants. Pediatr Res 62, 353–356 (2007). https://doi.org/10.1203/PDR.0b013e318123f719
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e318123f719
This article is cited by
-
Is there a relationship between dietary MSG obesity in animals or humans?
Amino Acids (2014)
-
Glutarazidurie Typ I
Monatsschrift Kinderheilkunde (2011)
