
Abstract
Isolated methylmalonic acidurias comprise a heterogeneous group of inborn errors of metabolism caused by defects of methylmalonyl-CoA mutase (MCM) (mut0, mut–) or deficient synthesis of its cofactor 5′-deoxyadenosylcobalamin (AdoCbl) (cblA, cblB). The aim of this study was to compare the long-term outcome in patients from these four enzymatic subgroups. Eighty-three patients with isolated methylmalonic acidurias (age 7–33 y) born between 1971 and 1997 were enzymatically characterized and prospectively followed to evaluate the long-term outcome (median follow-up period, 18 y). Patients with mut0 (n = 42), mut− (n = 10), cblA (n = 20), and cblB (n = 11) defects were included into the study. Thirty patients (37%) died, and 26 patients survived with a severe or moderate neurologic handicap (31%), whereas 27 patients (32%) remained neurologically uncompromised. Chronic renal failure (CRF) was found most frequently in mut0 (61%) and cblB patients (66%), and was predicted by the urinary excretion of methylmalonic acid (MMA) before CRF. Overall, patients with mut0 and cblB defects had an earlier onset of symptoms, a higher frequency of complications and deaths, and a more pronounced urinary excretion of MMA than those with mut− and cblA defects. In addition, long-term outcome was dependent on the age cohort and cobalamin responsiveness.
Similar content being viewed by others


Main
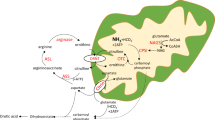
Isolated methylmalonic acidurias (synonym: methylmalonic acidemias) comprise a group of autosomal recessively inherited disorders characterized by an accumulation of MMA. They are caused by deficient activity of the homodimeric mitochondrial enzyme MCM (EC 5.4.99.2), which converts methylmalonyl-CoA to succinyl-CoA within the final catabolic pathway of l-isoleucine, l-valine, l-methionine, and l-threonine, odd-chain fatty acids, and the side chain of cholesterol. Deficient activity of MCM is caused by defects in the MCM apoenzyme or deficient intracellular synthesis of its cofactor AdoCbl. Two subgroups of MCM apoenzyme deficiency have been delineated, i.e. mut− defect with residual activity in the presence of high concentrations of AdoCbl and mut0 defect with complete loss of MCM activity (1). Both defects are caused by mutations in the MUT gene and therefore belong to the same complementation group (MIM #251000). Using somatic complementation analysis, defects of AdoCbl synthesis can be subdivided into several groups, three of which cause isolated methylmalonic acidurias, i.e. cblA, cblB, and, less frequently, cblD defects (2). The cblA defect (MIM #251100) is caused by mutations in the MMAA gene encoding a protein of unknown function (3), whereas the cblB defect (MIM #251110) is caused by mutations in the MMAB gene encoding cobalamin adenosyltransferase (EC 2.5.1.7) (4).
The clinical presentation of patients with isolated methylmalonic acidurias is variable; however, neonatal manifestation is usually associated with a severe disease course (5). Children with neonatal onset frequently present with recurrent vomiting, muscular hypotonia, progressive alteration of consciousness, and, finally, overwhelming illness that can progress to coma and death. Severe ketoacidosis and lactic acidosis, hyperammonemia, hyperglycemia, hypoglycemia, neutropenia, anemia, or pancytopenia are frequently found. If untreated, recurrent metabolic crises usually result in multiorgan failure or death. These metabolic crises are precipitated by conditions that are likely to induce catabolic state (e.g. febrile illness). In other patients with chronic progressive disease, psychomotor retardation and failure to thrive are the leading symptoms (6,7).
The first study on the natural history of methylmalonic aciduria demonstrated differences in the disease course and outcome of patients with isolated methylmalonic acidurias, with mut0 patients being most severely affected (8). However, this has not been studied in further detail in later studies, which have classified patients according to the onset of clinical symptoms and cobalamin responsiveness but not based on the underlying enzymatic defect (7,9,10). These studies have shown that the outcome was unfavorable in patients with early onset of symptoms and in those not responding to cobalamin. Although the overall survival has improved during the past two decades, the long-term outcome still remains disappointing (7,9). Neurologic outcome is often impaired by the manifestation of extrapyramidal movement disorder and developmental delay (10). Furthermore, CRF is frequently found (11). The major aim of this study was to investigate the long-term outcome in patients with isolated methylmalonic acidurias and whether the underlying enzymatic defect (mut0, mut−, cblA, and cblB) plays an important role in the outcome.
METHODS
Study population.
Eighty-three patients (45 female, 38 male) with a confirmed diagnosis of isolated methylmalonic acidurias from 37 European hospitals were included into this study. Patients were divided in three birth cohorts, i.e. 1970–1979 (cohort I), 1980–1989 (cohort II), and 1990–1997 (cohort III), and were followed until July 2004 (Table 1). Preliminary data on the follow-up of some patients until 1992 were reported more than one decade ago but did not differentiate between cblA and cblB defects (11).
The ethnic origin of the study patients was heterogeneous; however, most of them came from European countries, such as Germany (n = 26), Turkey (n = 13), Switzerland (n = 9), Belgium (n = 8), Italy (n = 8), Austria (n = 5), the Netherlands (n = 4), France (n = 2), Portugal (n = 2), and Greece (n = 1). Single patients from Tunisia, Libya, and Iran, or of unknown origin (n = 2) were also included. The frequency of known consanguinity in these families was high (30%; 22/73 families). Eighteen patients were siblings (nine families).
All mut0 and cblB patients received a low-protein diet (with or without administration of isoleucine-, methionine-, threonine-, and valine-free amino acid supplements); oral carnitine supplementation; and, in some cases, intermittent intestinal decontamination using metronidazole for maintenance treatment. Cobalamin-responsive patients were treated with hydroxycobalamin (OH-Cbl) or cyanocobalamin (i.m. or p.o.). Emergency treatment was performed during conditions precipitating a catabolic state (e.g. febrile illness, immunization, surgery). Two mut0 patients underwent orthotopic liver transplantation, one at the age of 17 mo and the other at the age of 8 y. These patients were followed separately after transplantation. Informed consent was obtained from patients and/or parents. The study was approved by the Institutional Review Board of the University of Basel, Switzerland.
Enzymatic classification of patients (mut0, mut−, cb1A, and cb1B defects).
Patients were classified as having mut0, mut−, cblA, or cblB defects by enzyme studies in cultured fibroblasts including analysis of MCM activity in crude cell or tissue homogenates (12), and incorporation of 14C-propionate into acid-precipitable materials in intact fibroblasts that were grown in basal medium with or without added OH-Cbl (1 or 10 μg/mL) as previously described (13,14). Both holo-MCM (assay without AdoCbl) and total MCM (assay with 50–100 μM AdoCbl) activities were strongly decreased in mut0 and mut− patients, whereas total MCM activity was normal in patients with deficient AdoCbl synthesis. Propionate incorporation was deficient in cells cultivated in basal medium, but cells showed a variable degree of response after administration of OH-Cbl, and, thus, in vitro OH-Cbl responsiveness was used to differentiate between mut0 and mut− patients. Cells of patients showing an at least 1.5-fold increase in propionate incorporation after administration of OH-Cbl were classified as mut−, whereas those with less or no increase were classified as mut0. Patients with deficient AdoCbl synthesis were differentiated into cblA defect using somatic complementation analysis or cblB defect using complementation analysis or analysis of cobalamin adenosyltransferase activity in fibroblast homogenates as described (2,15). In two patients, the enzymatic classification was based on previous results of an affected sibling.
There is no common consensus on the precise procedure for testing and evaluating cobalamin responsiveness. In vivo cobalamin responsiveness was usually tested by measurement of urinary MMA after repetitive administration of OH-Cbl (1–10 mg i.m. or i.v.) during a clinical and metabolic stable condition. In a minor subset of patients, a clinical trial of cobalamin was performed before stable conditions were achieved. However, most of these were retested later.
Urinary excretion of MMA.
Urinary concentration of MMA was quantified by gas chromatography/mass spectrometry. To avoid a bias induced by episodes of acute metabolic decompensation during which metabolic tests are more frequently performed than during episodes of compensated metabolic state, mean monthly values of urinary MMA excretion were calculated and used for the statistical analysis. Gas chromatography/mass spectrometry analyses were only included if performed before the manifestation of CRF.
Anthropometrics and assessment of neurologic and renal outcome.
A standardized questionnaire was sent out to referring physicians at three defined time points, i.e. at diagnosis, 1992, and 2003. The questionnaire asked for clinical presentation and major complications (e.g. metabolic stroke, CRF, and pancreatitis), anthropometrics, blood pressure, routine laboratory tests (including complete blood cell count, serum creatinine, serum uric acid, and urinary MMA concentration), results of cranial magnetic resonance imaging (MRI), treatment protocol, and psychomotor development (including results of different standardized psychological tests). In addition, files were examined in detail by two of the coauthors.
SD scores for length and head circumference were determined in comparison with the First Zurich Longitudinal Study of Growth and Development (16), and body weight was compared with age-specific percentiles from the same study (16).
Creatinine clearance was calculated from simultaneously obtained values of serum creatinine and supine length using the Schwartz formula (17). A creatinine clearance <60 mL/min/1.73 m2 was defined as CRF according to the European definition. Renal function was not evaluated in patients from cohort 1, who died before the age of 2 y because CRF was not recognized as a frequent complication at that time.
Statistical analysis.
Statistical analysis was performed using R (18). Nonparametric tests were used to calculate the differences in morbidity (renal complications and anthropometric changes) and mortality among age cohorts and enzymatically defined subgroups. Survival rates were compared by log-rank tests and were presented as Kaplan-Maier estimate survival curves. Onset of symptoms was tested for significant differences between groups using Kruskal-Wallis rank sum test. The Kruskal-Wallis test was also used to test the differences between medians of urinary MMA concentrations in enzymatically defined subgroups and the Wilcoxon rank sum test with continuity correction was used to test the differences in MMA excretion between patients with and without chronic renal failure. The incidence of chronic renal failure was tested for significant differences between groups using Pearson's χ2 test.
RESULTS
Enzymatic classification of patients.
Of the 83 patients, 42 (41 families) were classified as mut0, 10 (eight families) as mut−, 20 (15 families) as cblA, and 11 (nine families) as cblB patients. The median follow-up period was 18 y (range, 7–33 y; Table 1).
Cobalamin responsiveness.
In general, cobalamin responsiveness is difficult to evaluate, particularly in patients with relatively low MMA excretion. Among the well-documented patients, one of five tested cblB patients, three of four tested mut− patients, and all cblA patients responded to cobalamin therapy requiring variable doses (1 mg/d p.o. to 10 mg OH-Cbl i.m. twice weekly). In four additional mut− patients, a clinical trial with cobalamin was performed shortly after diagnosis. Three of them were reported to show a positive response (MMA data were not given); however, cobalamin therapy was stopped later because of noncompliance. The fourth patient was judged as nonresponsive by the responsible physicians. Of the two remaining mut− patients, one died in infancy before diagnosis and cobalamin was never tried in the other patient.
Urinary MMA excretion.
Serial investigations of urinary MMA concentrations (median number of investigations, 35; range, 15–207) were performed in 35 patients. They differed significantly between the enzymatic subgroups (Kruskal-Wallis rank sum test χ2 [3] = 665, p < 0.001), due to inter- and intraindividual variations there was considerable overlap (Fig. 1). Highest median concentrations were found in mut0 patients and lowest in cblA and mut− patients, whereas cblB patients had intermediate urinary MMA concentrations. Age distribution of patients at last investigation of urinary MMA did not differ between subgroups (Kruskal-Wallis rank sum test χ2 [3] = 6.13, p = 0.11).

Mean monthly urinary MMA concentrations in enzymatic subgroups. The defect groups included 14 mut0, three mut−, 11 cblA, and seven cblB patients. Mean monthly urinary MMA concentrations are shown as box plots.
Age at onset of symptoms and mortality.
Age at onset and disease progression differed between the four enzymatic subgroups. Earliest onset of symptoms was found in mut0 patients, whereas mut− patients showed latest onset of symptoms (Fig. 2). Forty-eight percent of patients developed symptoms within the first week of life, which was most common in mut0 patients (30/41 patients; median age at onset of symptoms, 5 d). The majority of these mut0 patients (25/30) had acute metabolic decompensation. Neonatal onset was found in 60% of patients with cblB (6/10; median age, 10 d), 55% of cblA patients (10/18; median age, 25 d), and 37% of mut− patients (3/8; median age, 75 d). In six patients, the onset of symptoms has not been documented. The differences in the onset of symptoms between groups showed a clear tendency, but did not reach statistical significance (Kruskal-Wallis rank sum test χ2 [3] = 7.54, p = 0.06).

Age at onset of symptoms in enzymatic subgroups of isolated methylmalonic acidurias. Age at onset of symptoms in 77 patients (41 mut0, eight mut−, 18 cblA, and 10 cblB patients) is shown as box plots.
Thirty patients (37%) died (median age, 2.7 y; range, 4 d to 22.6 y) with apparent differences in the frequency of death between the four enzymatic subgroups (log-rank test χ2 [3] = 11.2, p = 0.01; Fig. 3). Mortality was high in mut0 (20 deaths; median age at death, 2 y; range, 4 d to 22.6 y), cblB (five deaths; median age at death, 2.9 y; range, 7 d to 5.9 y), and mut− patients (four deaths; median age at death, 4.5 y; range, 4.6 mo to 8.9 y), but was less frequent in cblA patients (one death; age at death, 14 d). Furthermore, the survival rate of mut0 patients increased from the older (cohort I) to the younger (cohort III) birth cohorts (log-rank test χ2 [3] = 16.2, p < 0.005; Fig. 4) but remained unchanged for mut−, cblA, and cblB patients. Most deaths occurred during severe metabolic crises, but autopsies have only been performed rarely. One patient died unexpectedly due to cardiac arrest without previous cardiomyopathy and after full recovery from metabolic crisis.

Survival rate in patients from enzymatically defined groups. Survival rates are shown as Kaplan-Maier curves.

Survival rate in mut0 patients from different age cohorts. Survival is presented as Kaplan-Maier curves in the age cohorts I (year of birth, 1970–1979), II (year of birth, 1980–1989), and III (year of birth, 1990–1999).
Anthropometrics.
In general, growth retardation, failure to thrive, and mild microcephaly were frequently found in affected patients but again revealed some differences between subgroups. Failure to thrive was most frequently found in mut0 patients (47%), whereas it was rarely reported in cblA patients (5%). The median length was below the age-specific mean in all patients, but differed significantly between subgroups (Kruskal-Wallis rank sum test χ2 [3] = 9.12, p = 0.02). Again, mut0 patients (SD = −2.2; range, −6.2 to 0.4) were more severely affected than cblB (SD = −1.7; range, −4.9 to 1.8), mut− (SD = −1.05; range, −3.1 to 0.9) and cblA (SD = −1.0; range, −2.1 to 1). Mild microcephaly was quite common, but head circumference was not significantly different between subgroups (Kruskal-Wallis rank sum test χ2 [3] = 2.40, p = 0.49).
Renal complications.
CRF occurred in 43% of patients older than 2 y of age. The frequency of CRF differed between the four enzymatic subgroups (Pearson's χ2 test χ2 [3] = 12.82, p = 0.005). CRF was common in mut0 (61%) and cblB (66%) patients but was less frequently (cblA, 21%) or even not (mut–) found in other patients (Table 2). Median age at manifestation of CRF was 8 y (range, 2–18 y) in mut0, 11 y (range, 6.4–12 y) in cblA, and 13.5 y (range, 12–15 y) in cblB patients. Eight patients with CRF (four mut0, three cblA, one cblB) required hemodialysis. Secondary complications of CRF, such as anemia, arterial hypertension, and renal osteopathy further aggravated the disease course in these patients. Notably, the monthly mean excretion of MMA correlated with the occurrence of CRF (Wilcoxon rank sum test with continuity correction, W = 59307; p < 0.001; Fig. 5), suggesting that the amount of urinary MMA excretion may predict the occurrence of CRF.

Urinary MMA concentrations in patients with and without CRF. Mean monthly urinary MMA concentrations of patients with [n = 13; median age at last investigation; 9.2 (range, 2.1–21.4)] and without [n = 22; median age at last investigation, 15.9 (range, 2.9–28.1)] CRF are shown as box plots.
Both patients who underwent liver transplantation subsequently developed impaired renal function. One patient, who received a transplant at 18 mo of age, had CRF at age 12 y (P. Goyens and C. De Laet, personal communication), whereas the other patient who received a transplant at age 8 y had a decreased glomerular filtration rate but no CRF by age 14 y (19).
Neurologic complications.
Neurologic outcome was documented in 68 patients (78%). In general, the frequency of neurologic complications was high in all subgroups. Movement disorders (30%), seizures (23%), and mental retardation (25%) were the major complications. Psychosis and optic nerve atrophy were observed in a single patient. Cognitive dysfunction varied considerably between the subgroups. Again, mut0 patients had the poorest outcome (Fig. 6).

Cognitive function in patients from enzymatically defined groups. Patients in each defect group were divided in three groups based on their IQ or type of schooling. The defect groups included 24 mut0, five mut−, 19 cblA, and six cblB patients. White bars refer to patients with an IQ <60 or no schooling, gray bars to patients with an IQ between 60–90 or special schooling, and black bars to patients with an IQ >90 or normal schooling.
MRI studies in mut0 patients (n = 10) showed persistent abnormalities in seven patients, including basal ganglia lesions (n = 2), cortical atrophy (n = 3), and a more complex pattern in one patient presenting with arachnoidal cysts, Dandy-Walker malformation, and cerebellar hypoplasia. MRI studies in cblA patients (n = 8) showed cortical atrophy (n = 3) and white matter disease (n = 2) but no basal ganglia lesions. MRI scans in two mut− patients and one cblB patient revealed no abnormalities, whereas another cblB patient and one mut− patient had basal ganglia lesions.
Gastrointestinal complications.
Acute pancreatitis was reported in two mut0 patients resulting in insulin-dependent diabetes mellitus or death. Feeding difficulties and anorexia were frequently found and necessitated tube feeding in 13 patients, 11 of them belonging to the mut0 subgroup.
DISCUSSION
This study demonstrates that the long-term outcome in isolated methylmalonic acidurias is influenced by the underlying enzymatic defect (mut0, mut−, cblA, cblB). Earlier reports focused on the onset of symptoms and cobalamin responsiveness to classify the patients and to predict outcome (9,10). However, these parameters are variable and not sufficiently discriminative. Although previous studies have hypothesized that the enzymatic classification is a valuable tool for the assessment of outcome parameters (8,11), our study adds important new information on long-term follow-up and renal function.
In agreement with the observation of Matsui et al. (8), our study demonstrates that mut0 and cblB patients have a higher frequency of morbidity, mortality, and neurologic complications than mut– and cblA patients. In addition, as a result of the longer observation period, the present study adds the complication of chronic renal damage. A better neurologic outcome in mut– patients was also reported by Shevell et al. (20). However, none of their nine patients died or had an early onset of symptoms in contrast to our study and that of Matsui et al. Our results thus confirm the previous suggestion that a correct enzymatic classification of patients with isolated methylmalonic acidurias (mut0, mut−, cblA, and cblB) is helpful in predicting the long-term outcome and to specify the management of different patient groups.
The increased survival rate from the older to the younger birth cohorts, in particular in mut0 patients, mainly reflects an improvement of maintenance and emergency treatment strategies, as well as of neonatal care, diagnostics and increasing awareness of pediatricians for inborn errors of metabolism. It remains to be elucidated whether neonatal screening programs using tandem mass spectrometry are helpful in preventing the manifestation of neonatal crises and improving the outcome in methylmalonic acidurias. Because a considerable number of patients (in particular mut0) present clinically before a positive screening result is available, it has been questioned whether an inclusion of methylmalonic aciduria to neonatal screening programs would be successful (21). The present study demonstrates a large variation in the onset of symptoms (in particular, in mut−, cblA, and cblB patients), supporting that neonatal screening might be beneficial for these patients. Additionally, our observation in siblings from three families with a cblA defect provides evidence that especially milder forms of the disorder may profit from neonatal screening. The IQs of the three patients treated from the newborn period differed considerably from those of their older siblings (107 versus 78, 117 versus 73, and 120 versus 106).
Despite improving diagnostic workup and therapy, our study clearly shows that methylmalonic acidurias remain life threatening with a high burden of neurologic, renal, and other complications, in mut0 patients in particular. In addition, this study highlights that the outcome may also be severely compromised in the cblB defect, whereas long-term complications are less frequently found in mut− and cblA patients. It remains to be elucidated whether this difference is related to cobalamin responsiveness, which was often found in mut− and always in cblA patients but only rarely in cblB patients and never found in mut0 patients in our study. Cobalamin responsiveness should be tested carefully in each patient, particularly in cblB patients to avoid misclassification of patients with a mild response. Unfortunately, in vitro cobalamin responsiveness does not reliably predict in vivo responsiveness (13), and standardized protocols to test cobalamin responsiveness have not yet been established.
Serial analyses of urinary MMA concentrations reveal significant differences among the four enzymatic subgroups. This is not unexpected because the severity of MCM deficiency and the degree of cobalamin responsiveness influence the accumulation of MMA. However, urinary MMA excretion has a considerable day-to-day variation in individual patients, mainly influenced by fasting, intake of natural protein, and hydration (22). Furthermore, the collection of serial urine samples was not standardized with regard to nutritional parameters and thus is a source of bias. Nevertheless, we demonstrate a significant difference in the urinary concentration of MMA between the subgroups, suggesting that this is a robust follow-up parameter before onset of CRF. Interestingly, the highest mean concentrations of MMA were found in patients who later developed CRF. In this light, an estimation of cumulative MMA excretion over time may be helpful to assess the risk of CRF. This notion is supported by our finding that CRF was not found in any patient with urinary MMA excretion below an approximate threshold of 2000 mmol/mol creatinine, even in patients older than 20 y. However, we cannot exclude that even these patients will develop CRF over a longer time period. Taken together, the age at onset and progression of CRF seem to depend on the severity of the disorder as predicted by urinary MMA excretion. Of course, these results require confirmation from prospective follow-up studies with standardized biochemical monitoring. Furthermore, it remains to be determined whether blood MMA and/or propionylcarnitine levels may also predict the outcome.
The spectrum of neurologic complications observed in mut0 and cblB patients is in accordance with a previous study in a cohort of cobalamin nonresponders with an early onset of symptoms (10). However, in the present study, we demonstrate that a minority of mut0 patients remained neurologically unaffected. It is unknown whether this reflects the beneficial effects of therapy or is influenced by individual protecting factors modifying the natural history of the disease. Although many variables have been considered, we cannot exclude a confounder bias caused by noncompliance or other factors that might have influenced the outcome. The pathophysiological basis for the manifestation of cerebral symptoms and renal failure is not yet fully understood; however, some studies have shown that organic acids and CoA esters that accumulate in methylmalonic acidurias (MMA, 2-methylcitrate, and propionyl-CoA) induce a synergistic inhibition of mitochondrial energy metabolism (23,24). In line with this, we previously hypothesized that MMA may act as a nephrotoxin in concert with other metabolites (25) and reported on a mut0 patient with end-stage renal failure who improved after reinforcement of dietary treatment (26).
In conclusion, our study demonstrates that the natural history and outcome of MMAs is considerably influenced by the underlying defect linking the biochemical to the clinical phenotype. This hypothesis should be tested using an international prospective multicenter study.
Abbreviations
- AdoCbl:
-
5′-deoxyadenosylcobalamin
- CRF:
-
chronic renal failure
- MCM:
-
methylmalonyl-CoA mutase (EC 5.4.99.2)
- MMA:
-
methylmalonic acid
- OH-Cbl:
-
hydroxycobalamin
References
Willard HF, Rosenberg LE 1980 Inherited methylmalonyl CoA mutase apoenzyme deficiency in human fibroblasts. Evidence for allelic heterogeneity, genetic compounds, and codominant expression. J Clin Invest 65: 690–698
Suormala T, Baumgartner MR, Coelho D, Zavadakova P, Kozich V, Koch HG, Berghauser M, Wreight JE, Burlina A, Sewell A, Herwig J, Fowler B 2004 The cblD defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J Biol Chem 279: 42742–42749
Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dorè C, Hudson T, Rosenblatt DS, Gravel RA 2002 Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci U S A 99: 15554–15559
Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA 2002 Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet 11: 3361–3369
Fenton WA, Gravel RA, Rosenblatt DS 2001 Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill, New York, pp 2165–2193
Hörster F, Hoffmann GF 2004 Pathophysiology, diagnosis, and treatment of methylmalonic aciduria-recent advances and new challenges. Pediatr Nephrol 19: 1071–1074
de Baulny HO, Benoist JF, Rigal O, Touati G, Rabier D, Saudubray JM 2005 Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis 28: 415–423
Matsui SM, Mahoney MJ, Rosenberg LE 1983 The natural history of the inherited methylmalonic acidemias. N Engl J Med 308: 857–861
Van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C 1994 Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr 125: 903–908
Nicolaides P, Leonard JV, Surtees R 1998 The neurological outcome of methylmalonic acidaemia. Arch Dis Child 78: 508–512
Baumgarter ER, Viardot C 1995 Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. J Inherit Metab Dis 18: 138–142
Baumgartner ER 1983 Activity of the cobalamin-dependent methylmalonyl-CoA mutase. In: Hall CA (ed) The Cobalamins: Methods in Hematology. Churchill Livingstone, Edinburgh, New York, pp 181–195
Baumgartner R, Giardini O, Cantani A, Sabetta G, Castro M 1982 Methylmalonic acidaemia due to mutase apoenzyme defect: responsive to vitamin B12 in intact fibroblasts but not in vivo. J Inherit Metab Dis 5: 137–141
Willard HF, Ambani LM, Hart AC, Mahoney MJ, Rosenberg LE 1976 Rapid prenatal and postnatal detection of inborn errors of propionate, methylmalonate and cobalamin metabolism- a sensitive assay using cultured cells. Hum Genet 34: 277–283
Fenton WA, Rosenberg LE 1981 The defect in the cblB class of human methylmalonic acidemia: deficiency of cob(I)alamin adenosyltransferase activity in extracts of cultured fibroblasts. Biochem Biophys Res Commun 98: 283–289
Prader A, Largo RH, Molinari L, Issler C 1989 Physical growth of Swiss children from birth to 20 years of age. First Zurich longitudinal study of growth and development. Helv Paediatr Acta Suppl 52: 1–125
Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A 1976 A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics 58: 259–263
R: a language and environment for statistical computing. Available at: http://www.R-project.org. Accessed June 01, 2005
Fuoti M, Pinotti M, Villa MC, celano MR, Miceli V, Amoruso C, Rossi G, Parini R, Nebbia G 2005 Hepatic transplantation in methylmalonic acidemia: a case report. Acta Gastroenterol Belg 68: 491
Shevell MI, Matiaszuk N, Ledley FD, Rosenblatt DS 1993 Varying neurological phenotypes among mut0 and mut- patients with methylmalonyl CoA mutase deficiency. Am J Med Genet 45: 619–624
Leonard JV, Vijayaraghavan S, Walter JH 2003 The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr 162: S21–S24
Thompson GN, Chalmers RA 1990 Increased urinary metabolite excretion during fasting in disorders of propionate metabolism. Pediatr Res 27: 413–418
Brock M, Buckel W 2004 On the mechanism of action of the antifungal agent propionate. Eur J Biochem 271: 3227–3241
Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, van den Heuvel LP, Drose S, Brandt U, Hoffmann GF, Ter Laak H, Kölker S, Smeitink JA 2006 Secondary mitochondrial dysfunction in propionic aciduria, a pathogenic role for endogenous mitochondrialtoxins. Biochem J 398: 107–112
Kölker S, Okun JG 2005 Methylmalonic acid—an endogenous toxin?. Cell Mol Life Sci 62: 621–624
Schmitt CP, Mehls O, Trefz FK, Hörster F, Weber TL, Kölker S 2004 Reversible end-stage renal disease in an adolescent patient with methylmalonic aciduria. Pediatr Nephrol 19: 1182–1184
Acknowledgements
This study is dedicated to the patients and their families whom the authors thank for their trust and kind cooperation. They thank Mira Günther for her excellent technical assistance with enzymatic studies. They are most thankful for clinical information on patients, which was kindly provided by the following colleagues: C. Boujet, A. Burtscher, I. Braun, F. Carnevale, C. De Laet, C. Dionisi-Vici, F.J.M. Eyskens, B. Francois, F. Freundl, P. Goyens, S. Grünewald, J. Hennermann, B. Hoppe, S. Huber, J. Jaeken, A. Jonnard, H. Korall, I. Kreuder, M. Leichsenring, D. Leupold, M. Lindner, H. Machnik, W. Marg, I. Marquardt, K. Michalski, E. Mönch, D. Möslinger, H. Niederhoff, J.M. Nuoffer, R. Parini, B. Plecko, M.E. Rubio, F. Sarrot-Reynaud, S. Scheibenreiter, D. Scheible, H. Schmidt, D. Schneider, C. Schröder, M. Schütte de Lujan, O. Schwab, E. Simon, K.F. Trefz, S. van der Meer, U. Wendel, and F.A. Wijburg.
Author information
Authors and Affiliations
Additional information
This study was supported by Deutsche Forschungsgemeinschaft (DFG grant HO2501/1-1 to F. Hörster), the Swiss National Science Foundation (grant 3200-066878 to B. Fowler, T. Suormala, and M.R. Baumgartner) and the German Federal Ministry of Education and Science (BMBF grant 01GM0305 to P. Burgard).
ArticlePlus
Click on the links below to access all the ArticlePlus for this article.
Please note that ArticlePlus files may launch a viewer application outside of your web browser.
Rights and permissions
About this article
Cite this article
Hörster, F., Baumgartner, M., Viardot, C. et al. Long-Term Outcome in Methylmalonic Acidurias Is Influenced by the Underlying Defect (mut0, mut−, cblA, cblB). Pediatr Res 62, 225–230 (2007). https://doi.org/10.1203/PDR.0b013e3180a0325f
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3180a0325f
This article is cited by
-
The impact of metabolic stressors on mitochondrial homeostasis in a renal epithelial cell model of methylmalonic aciduria
Scientific Reports (2023)
-
Clinical and Molecular Spectrum of Patients with Methylmalonic Acidemia
Indian Journal of Pediatrics (2023)
-
Methylmalonic Acid Impairs Cell Respiration and Glutamate Uptake in C6 Rat Glioma Cells: Implications for Methylmalonic Acidemia
Cellular and Molecular Neurobiology (2023)
-
Spectrum and characterization of bi-allelic variants in MMAB causing cblB-type methylmalonic aciduria
Human Genetics (2022)
-
Review of neuropsychological outcomes in isolated methylmalonic acidemia: recommendations for assessing impact of treatments
Metabolic Brain Disease (2022)
