
Abstract
Meconium aspiration syndrome is a serious condition of the newborn characterized by pulmonary inflammation with substantial neutrophil infiltration. We recently showed that meconium is a potent activator of complement. The aim of the present study was to investigate a possible role for complement in meconium-induced neutrophil activation. Meconium was incubated in human whole blood anticoagulated with lepirudin, a specific thrombin inhibitor that does not affect complement activation. Complement activation was detected by measuring the terminal complement complex. Neutrophil oxidative burst and changes in CD11b and l-selectin expression were measured by flow cytometry. Complement was inhibited using the MAb 166-32 and 137-26, which block factor D and neutralize C5a, respectively. Meconium markedly activated the neutrophils, as revealed by up-regulation of CD11b, accentuation of l-selectin shedding, and induction of oxidative burst. Complement inhibition using the anti–factor D antibody completely (95–100%) blocked meconium-induced changes in CD11b and l-selectin expression, whereas oxidative burst was reduced by 60–70%. The anti-C5a antibody inhibited the neutrophil activation to the same extent as anti–factor D. The data suggest that complement activation is largely responsible for the neutrophil inflammatory responses induced by meconium in vitro and that C5a is a key mediator of this response.
Similar content being viewed by others

Main
Meconium aspiration syndrome (MAS) may cause a profound lung inflammation similar to acute respiratory distress syndrome (ARDS). A substantial increase in neutrophils in the airways; increased levels of inflammatory cytokines, eicosanoids, superoxide anions, and endothelin-1; expression of inducible nitric oxide synthase and cyclooxygenase-2; and activation of nuclear factor-κB have been described in MAS (1–8). A profound inflammatory response and the release of leukocyte cytotoxic products have an important role in initiation and propagation of lung injury in ARDS, in which neutrophils and their cytotoxic products play a crucial role (9).
The complement system is a key mediator of inflammation. Complement-mediated activation of neutrophils and monocytes leads to cytokine production; release of lipid mediators and histamine; oxidative burst; and altered expression of adhesion molecules, including increased expression of CD11b and shedding, revealed as decreased expression of CD62L. We showed recently that meconium is a potent activator of the alternative complement pathway in vitro (10). To study the interaction between all participating inflammatory systems, we used an in vitro human whole-blood model that is based on the use of lepirudin, a thrombin-specific anticoagulant that does not interfere with complement activation (11). We hypothesized that meconium-induced complement activation in whole blood would lead to neutrophil activation and examined the effect of meconium and inhibition of complement on neutrophil oxidative burst and surface expression of adhesion molecules CD11b and CD62L (l-selectin).
METHODS
Equipment and reagents.
All tips and tubes used in these experiments were endotoxin-free. Polypropylene tubes (Falcon; Becton Dickinson, Franklin Lakes, NJ) were used to obtain low background activation of complement. Nunc cryotubes (Nalgene; NUNC, Roskilde, Denmark) were used for blood collection and freezing. Sterile PBS was from Invitrogen (Paisley, Scotland), Lepirudin (Refludan) was from Hoechst (Frankfurt am Main, Germany), and EDTA (E-4884) and DMSO (D-2650) were from Sigma Chemical Co.-Aldrich (Schnelldorf, Germany). The following mouse anti-human phycoerythrin (PE)-conjugated MAbs were used for detection of surface antigens: anti-CD11b (IgG2a, clone D12; BD Biosciences, San Jose, CA) and anti-CD62L (IgG1, clone DREG-56; BD Biosciences, Pharmingen, Belgium). Oxidative burst was measured by flow cytometry using dihydrorhodamine (DHR) 123 (F-37370, Fluka; Sigma Chemical Co.-Aldrich, Schnelldorf, Germany). Phorbol-12-β-myristat-13-α-acetate (PMA) (F-79350, Fluka; Sigma Chemical Co.-Aldrich) was used as positive control. Flow cytometry was performed with a FACScalibur (Becton Dickinson, San Jose, CA). OD from the ELISA experiments was read in an MRX microplate reader (Dynex Technologies, Dekendorf, Germany).
Complement inhibitory antibodies.
Mouse MAbs to human factor D (clone 166-32; IgG1), C5a (clone 137-26; IgG1), and the isotype-matched control (clone G3-519; anti–HIV-1 gp120) were produced and purified under identical conditions in the laboratory of one of the authors (M.F.). Anti-C5a 137-26 is highly specific for the C5a moiety on native C5 without interfering with C5 cleavage as described in detail previously (12). Thus, this antibody preneutralizes C5a and thereby blocks its biologic effects before it is released. The anti–factor D antibody is highly specific for factor D of the alternative complement pathway as documented in detail previously (13,14).
Meconium.
Samples of meconium from 54 healthy newborns were collected from diapers with a wooden spatula and stored in 50 mL of polypropylene centrifuge tubes (Corning, Acton, MA) at −20°C. Lipopolysaccharide (LPS) was quantified in all samples using the Limulus Ameboecyte Lysate Assay (LAL QCL-1000, 50-648U; BioWhittaker/Cambrex Bio Science, Walkersville, MD). Samples that contained >100 pg of LPS/mg of meconium were excluded, and the remaining 42 samples were thawed, pooled, and processed by nonpyrogenic pipette (50 mL; Costar Corp., Cambridge, MA) in PBS and collected in surface bottles (NUNC Brand Production). The batch then was freeze-dried (HETO-FD3; Heto, Holten, Denmark). The weight difference between meconium and freeze-dried meconium indicated that the meconium water content was 75%. We reconstituted the meconium with the corresponding amount of distilled water giving a meconium concentration of ∼340 mg/mL. This preparation was diluted in PBS to a final concentration of 100 mg/mL, split in aliquots, and frozen at −20°C until the day of experiment.
Human whole blood.
The experiments with adult human whole blood were performed using samples from six healthy volunteers. Cord blood was obtained after normal deliveries of healthy newborns by healthy mothers. Informed consent was obtained from the parents. Three different cord blood samples were obtained for the surface marker and oxidative burst experiments, respectively.
Experimental model.
A recently developed whole-blood model to study the role of complement activation was used (11). The model is based on anticoagulation with lepirudin, a recombinant hirudin analogue that is a highly specific thrombin inhibitor that does not influence complement activation, in contrast to heparin. Whole blood was collected in polypropylene tubes that contained lepirudin (50 μg/mL) and preincubated with PBS or antibody for 5 min in a water bath at 37°C. Optimal experimental conditions were defined in pilot experiments using meconium concentrations that gave close to maximal signals in the different readouts and concentrations of inhibitors that gave maximal inhibition. Thus, we used 0.1 mg of meconium/mL and 10 μg of antibody/mL in the CD11b and CD62L experiments and 2.5 mg of meconium/mL and 50 μg of antibody/mL in the burst experiments. After preincubation, meconium was added and samples were incubated for another 30 min. After incubation, the samples were processed immediately in two different directions, one for flow cytometric detection and the other for detection of complement activation. EDTA was added to the latter to a final concentration of 20 mM. Plasma was obtained by centrifugation at 1000 × g for 8 min and immediately frozen and stored at −70°C until analysis.
CD11b and CD62L expression in neutrophils.
After incubation, cells were fixed with 0.5% (vol/vol) paraformaldehyde for 4 min in water bath at 37°C. The cells were stained with either anti–CD11b-PE or anti–CD62L-PE for 15 min, lysed, and washed before the median fluorescence intensity (MFI) was measured by flow cytometry.
Oxidative burst in neutrophils.
The stock solution of DHR 123 was 1 mg/mL in DMSO. It was stored at −20°C in small aliquots. Before the experiments, the stock was diluted with PBS to give a working solution of 50 μg/mL. The final concentration in the samples was 6.25 μg/mL. PMA was solved in DMSO to a stock solution of 1 mg/mL and stored at −20°C in small aliquots. Before the experiments, the PMA stock was diluted with PBS to give a working solution of 2 μg/mL (final concentration in the samples was 0.29 μg/mL) and was added to the tube at the time of preincubation. After activation with meconium, DHR was added, and the samples were incubated for another 10 min in water bath at 37°C. Then the cells were lysed and washed before MFI was measured by flow cytometry.
Complement activation.
Activation of complement was measured by quantifying the soluble terminal sC5b-9 complement complex (TCC) using an ELISA based on the MAb aE11 specific for a neoepitope exposed in C9 when incorporated in TCC, principally as described previously (15).
Statistical analysis.
Data are presented as median values and total range (minimum-maximum). Figures show box and whiskers where the central box shows the data between quartiles with the median represented by the horizontal line and the vertical lines show minimum and maximum values. Wilcoxon signed ranks test was used to compare dependent groups (SPSS for windows, version 11.0; SPSS, Chicago, IL). The significance level was defined by p < 0.05.
RESULTS
Neutrophil Surface Markers
CD11b expression.
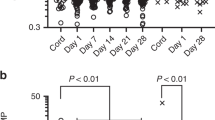
Meconium increased neutrophil CD11b expression to 299 MFI (279–556) compared with 151 MFI (39–466) for the PBS control (p < 0.05; Fig. 1). The meconium-induced CD11b increase was completely abolished by anti–factor D [157 MFI (128–330)] and by anti-C5a [83 MFI (19–251); p < 0.05]. No inhibition was seen for the isotype-matched control antibody [324 MFI (279–580)].

CD11b expression. Meconium (0.1 mg/mL; MEC) significantly (p < 0.05) increased neutrophil CD11b expression when incubated in human whole blood for 30 min. This up-regulation was completely abolished (p < 0.05) in the presence of 10 μg/mL of anti–factor D (MEC+aD) or 10 μg/mL of anti-C5a (MEC+aC5), whereas no effect was seen using an isotype-matched control antibody (10 μg/mL; MEC+Ctr). The data are median and range of six different experiments, each from a different blood donor (n = 6). T30, CD11b expression after 30 min in whole blood not incubated with meconium; *significantly different from T30; *significantly different from MEC.
CD62L expression.
Meconium enhanced neutrophil CD62 shedding to 3 MFI (2–7) compared with 12 MFI (4–84) for the PBS control (p < 0.05; Fig. 2). The meconium-induced CD62L shedding was completely abolished by anti–factor D to 20 MFI (6–49) and by anti-C5a to 35 MFI (9–130; p < 0.05). No inhibition of shedding was seen for the isotype-matched control antibody [4 MFI (2–53)].

CD62L expression. Meconium (0.1 mg/mL; MEC) significantly (p < 0.05) reduced neutrophil CD62L (l-selectin) expression when incubated in human whole blood for 30 min. This down-regulation was completely abolished (p < 0.05) in the presence of 10 μg/mL of anti–factor D (MEC+aD) or 10 μg/mL of anti-C5a (MEC+aC5), whereas no effect was seen using an isotype-matched control antibody (10 μg/mL; MEC+Ctr). The data are median and range of six different experiments, each from a different blood donor (n = 6). T30, CD62L expression after 30 min in whole blood not incubated with meconium; *significantly different from T30; **significantly different from MEC.
Complement activation in the surface marker experiments.
In the CD11b and CD62 experiments, the levels of TCC were measured in plasma. Meconium increased TCC formation to 18 arbitrary units (AU)/mL (13–38) compared with 1 (0–3) for the PBS control (p < 0.05; Fig. 3). The meconium-induced increase in TCC was completely abolished by anti–factor D [1 AU/mL (0.5–3)]. As expected, anti-C5a treatment did not reduce TCC formation because it does not block C5 cleavage. In contrast, it slightly but significantly increased TCC formation to 36 MFI (23–66; p < 0.05). No change in TCC formation was seen for the isotype-matched control antibody [19 MFI (13–38)].

TCC in CD11b and CD62L experiments. TCC was used as an indicator of complement activation and measured in plasma separated from the whole blood after incubation. Meconium (0.1 mg/mL; MEC) induced a significant (p < 0.05) increase in TCC in the CD11b and CD62L experiments described in Figures 1 and 2. Anti–factor D (10 μg/mL) completely inhibited (p < 0.05) meconium-induced complement activation (MEC+aD), whereas anti-C5a treatment (10 μg/mL) slightly and significantly (p < 0.05) increased TCC formation (MEC+aC5). No inhibition of complement activation was seen with the isotype-matched control antibody (10 μg/mL; MEC+Ctr). The data are median and range of six different experiments, each from a different blood donor (n = 6). T30, TCC concentration after 30 min incubation of whole blood not incubated with meconium; *significantly different from T30; **significantly different from MEC.
Neutrophil Oxidative Burst
Meconium induced oxidative burst to 60 MFI (27–98) compared with 0 MFI (0–0.3) for the PBS control (p < 0.05; Fig. 4). The meconium-induced oxidative burst was significantly inhibited by anti–factor D to 19 MFI (5–38) and by anti-C5a to 23 MFI (9–83), corresponding to 68 and 62% reduction, respectively (p < 0.05). No inhibition was seen for the isotype-matched control antibody [55 MFI (23–108)].

Neutrophil oxidative burst. Meconium (2.5 mg/mL; MEC) significantly (p < 0.05) increased neutrophil oxidative burst when incubated in human whole blood for 30 min. This increase was significantly (p < 0.05) reduced in the presence of 50 μg/mL of anti–factor D (MEC+aD) or 50 μg/mL of anti-C5a (MEC+aC5), whereas no effect was seen using an isotype-matched control antibody (50 μg/mL; MEC+Ctr). The data are median and range of six different experiments, each from a different blood donor (n = 6). T30, oxidative burst after 30 min in whole blood not incubated with meconium; *significantly different from T30; **significantly different from MEC.
Complement activation in the oxidative burst experiments.
In the oxidative burst experiments, the levels of TCC were measured in plasma. Meconium increased TCC formation to 44 AU/mL (30–50) compared with 2 AU/mL (0.7–3) for the PBS control (p < 0.05; Fig. 5). The meconium-increased TCC formation was virtually but not completely abolished by anti–factor D [7 AU/mL (4–10)]. As seen for the surface marker experiments, anti-C5a treatment slightly but significantly increased TCC formation to 58 AU/mL (43–84; p < 0.05). No change in TCC formation was seen for the isotype-matched control antibody [41 AU/mL (29–46)].

TCC in oxidative burst experiments. Meconium (2.5 mg/mL; MEC) induced a significant (p < 0.05) increase in TCC in the oxidative burst experiments described in Figure 4. Anti–factor D (50 μg/mL) virtually but not completely abolished (p < 0.05) the meconium-induced complement activation (MEC+aD), whereas anti-C5a treatment (50 μg/mL) slightly and significantly (p < 0.05) increased complement activation (MEC+aC5). No inhibition of complement activation was seen with the isotype-matched control antibody (50 μg/mL; MEC+Ctr). The data are median and range of six different experiments, each from a different blood donor (n = 6). T30, TCC concentration after 30 min incubation of whole blood not incubated with meconium; *significantly different from T30; **significantly different from MEC.
Cord Blood
CD11b.
Meconium (0.04–5.0 mg/mL) induced a dose-dependent increase in neutrophil CD11b expression in cord blood (Fig. 6A). Using 2.5 mg of meconium/mL of blood, this increase was dose-dependently inhibited by anti–factor D and anti-C5a (Fig. 6B). The dose of antibodies required for inhibition was higher (50 μg/mL) than that for adult blood (10 μg/mL), consistent with the higher dose of meconium needed for sufficient CD11b expression (2.5 mg/mL) compared with that used in adult blood (0.1 mg/mL).

CD11b and oxidative burst in cord blood. Meconium induced up-regulation of neutrophil CD11b expression in a dose-response manner (0.04–5 mg/mL) when incubated in human cord blood for 15 min (A). T15/T30, neutrophil CD11b expression in human cord blood not incubated with meconium after 15/30 min. Anti–factor D and anti-C5a dose-dependently (25 and 50 μg/mL, respectively) inhibited the meconium (2.5 mg/mL)-induced up-regulation of CD11b (B). Meconium induced increase in neutrophil oxidative burst in a dose-response manner (0.625–5 mg/mL) when incubated in human cord blood for 30 min (C). T30, neutrophil oxidative burst in human cord blood not incubated with meconium after 30 min. Anti–factor D and anti-C5a inhibited the meconium (2.5 mg/mL)-induced oxidative burst, the former equally effective and the latter dose-dependently using 25 and 50 μg/mL, respectively (D). Data from one representative of three experiments are shown.
Oxidative burst.
Meconium (0.625–5.0 mg/mL) induced a dose-dependent increase in neutrophil oxidative burst in cord blood (Fig. 6C). Using 2.5 mg of meconium/mL of blood, this increase was inhibited by anti–factor D comparable to the adult blood but required a higher concentration of anti-C5a (50 μg/mL) compared with adult blood (10 μg/mL; Fig. 6D).
CD62L.
Cord-blood CD62L was subjected to extensive spontaneous shedding from the neutrophils during the incubation period. Although detectable at the start of the incubation, it was completely shed in the PBS control during the incubation period, which made it impossible to investigate the effect of meconium on CD62L shedding; thus, inhibition of shedding with complement inhibitors could not be investigated.
DISCUSSION
Neutrophils are suggested to be of importance in development of meconium-induced lung inflammation (16). We showed recently that components of meconium present in both the lipid and water fractions of meconium are potent activators of complement in human serum and in piglets (10). Because complement activation, with release of activation products including the highly potent fragment C5a, is known to activate neutrophils, we hypothesized a possible role for complement in meconium-induced neutrophil activation. The present work for the first time demonstrates that complement activation is essential in meconium-induced neutrophil activation. CD11b, CD62L, and oxidative burst play important pathophysiologic roles in different diseases and are valuable markers of inflammation. The role of complement in leukocyte trafficking in the lungs and the interplay among complement, adhesion molecules, oxidative burst, and the cytokine and chemokine network in pulmonary inflammation was reviewed recently (17). Consistently, our data indicate that complement activation is upstream of the inflammatory cell activation because the specific complement inhibitors anti–factor D and anti-C5a markedly attenuated neutrophil activation. The two antibodies inhibited the neutrophil response to the same extent, indicating that the alternative complement pathway is essential and that the key effector molecule is C5a.
CD11b plays a central role in inflammation and in the systemic inflammatory response syndrome (18). Integrin belongs to a family of adhesion receptors and is found on the majority of cells in the body (19). Upon activation, the surface expression and binding affinity of CD11b are increased. Neutrophil adhesion to the endothelium is upstream of neutrophil-mediated endothelial cell damage, microvascular injury, and organ failure (20). In the present study, we show for the first time that meconium-induced up-regulation of CD11b is entirely complement dependent and abolished using complement-specific inhibitors.
l-selectin (CD62L) is an adhesion molecule that is expressed on the surface of leucocytes that are involved in the initial phase of the inflammatory response. l-selectin is an important early molecule involved in leukocyte-endothelial interactions. It is rapidly shed from neutrophils, monocytes, and lymphocytes during activation by different stimuli and during the rolling process, allowing the leukocyte to attach firmly to the endothelium by up-regulated CD11b (21). We found that l-selectin was markedly shed upon exposure of meconium to the blood. Complement inhibition efficiently abolished shedding of l-selectin from the leukocyte surface, indicating a central role for complement in the initial neutrophil inflammatory activation.
Oxidative burst, i.e. the production of reactive oxygen species, is an important defense mechanism by which phagocytes destroy micro-organisms (22–24). In a rat model of IgG immune complex–induced lung injury, reactive oxygen species may explain early induction of lung injury (within 30 min). The expression of inflammatory cytokines would not be expected during this short period of time, in contrast to complement activation, which is an early event (17). Reactive oxygen species cause tissue injury in local as well as systemic inflammatory conditions. Pulmonary or systemic insults may result in acute lung injury or ARDS, the most severe form of acute lung injury, both of which are associated with a high rate of mortality (25–28). Patients with ARDS have increased levels of H2O2 in their breath condensate (9,29,30). MAS may develop into ARDS, but data on the role of oxidative burst in MAS are somewhat conflicting (31–33). In the present study, we found that meconium is a strong inducer of neutrophil oxidative burst in vitro. The meconium dose needed to induce oxidative burst was substantially higher than what was required to get changes in CD11b and CD62L expression. Although we found a substantial reduction in oxidative burst by inhibition of complement, this inhibition was not complete, in contrast to what was found for the changes in CD11b and CD62L. This may suggest that the oxidative burst is partly dependent on other factors than complement or that complete inhibition of complement was more difficult to obtain when using the highest doses of meconium. The latter notion is supported by the TCC data, where an efficient but not complete inhibition of complement was obtained in the burst experiments (Fig. 5), compared with a complete inhibition in the CD11b/CD62L experiments (Fig. 3).
Treatment of MAS to date been has largely symptomatic (2,3,34–42). Complement inhibition has been suggested as a therapeutic alternative in several conditions in which the system has been documented to contribute to the pathogenesis of the disease (23,43–45). Our in vitro data suggest that complement may have a primary role in meconium-induced inflammation; thus, inhibition of complement should be considered as a possible therapeutic approach in MAS. We showed previously that meconium-induced complement activation is mediated by the alternative pathway. Furthermore, it is widely known that the interaction between C5a and its receptor, C5aR, is an essential mechanism in the development of an inflammatory response (46,47). Both anti–factor D, blocking the alternative pathway, and anti-C5a had similar effectiveness in inhibition of meconium-induced neutrophil inflammatory response, suggesting that both factor D and C5a are possible candidates for therapeutic inhibition.
For logistic reasons, it is much more difficult to obtain cord blood than adult blood for a study that is highly dependent on fresh whole blood for flow cytometry analysis. We therefore examined adult human blood extensively in our study and then performed similar experiments using a limited number of cord-blood samples. This was mandatory because cord and adult blood differ substantially in several manners. The time lag between the birth of the baby and the birth of placenta and the time needed for transport of cord blood to laboratory both may induce spontaneous activation of CD11b, oxidative burst, and spontaneous shedding of CD62L. These factors make in vitro experiments with cord blood less accurate in pathophysiologic studies of MAS compared with adult blood experiments, which can be performed under strictly controlled conditions. It was shown previously that cord-blood neutrophils have lower CD62L expression compared with adult blood (48,49). Our data are in accordance with these observations. In fact, the CD62L was completely shed spontaneously during the incubation period, making it impossible to study the effect of meconium on CD62L shedding. In contrast, CD11b expression and oxidative burst could reliably be examined in cord blood. Despite the need for a higher amount of meconium to induce CD11b in cord blood, the effect of meconium on CD11b and oxidative burst was similar to adult blood, as was the effect of complement inhibition. The low number of cord-blood experiments performed does not allow statistical testing. However, that we describe principally identical results for adult and cord blood, i.e. a substantial inhibitory effect using both complement inhibitory antibodies, and that the effect was dose dependent support the notion that the data obtained with adult blood are presumably valid for newborns as well.
Whereas the anti–factor D MAb efficiently inhibited TCC formation, consistent with blocking of the alternative complement pathway, the anti-C5a MAb neutralizes C5a without inhibition of complement activation. In fact, TCC formation was slightly enhanced in the presence of anti-C5a. This observation may seem surprising at first glance but is consistent with previous data obtained when using this antibody to inhibit bacteria-induced C5a effects (50). The increased TCC formation cannot be explained by immune-complex formation because we excluded this possibility by experiments performed when this antibody was characterized (12). The most likely explanation for this effect is that binding of the antibody to native C5 increases the susceptibility of C5 to cleavage by the C5 convertase. Irrespective of the explanation for this phenomenon, a theoretical advantage with this antibody is that the harmful effects of C5a are attenuated, whereas the bacteriolytic activity by C5b-9 is preserved.
LPS is a component of outer membrane of Gram-negative bacteria. It binds to CD14 and/or CD11b/CD18 at the cell surface and facilitates an interaction with Toll-like receptor 4-MD complex, which via signal transduction cascades ultimately leads to activation of transcription factors (51). The interplay between LPS and complement is complex (52,53). A possible effect of LPS in the meconium-induced inflammatory reaction should be considered. We therefore measured the amount of LPS in each meconium sample and included only those with pictogram levels in the batch. Although these low amounts may induce some cytokine synthesis, amount in the range of micrograms is needed for LPS to induce complement activation (54). Thus, it can virtually be excluded that LPS contributes to the complement-induced inflammatory reaction observed in the present study.
CONCLUSION
In summary, our in vitro data suggest that complement activation is upstream of the neutrophil inflammatory responses induced by meconium and that C5a is a key mediator of this response.
Abbreviations
- ARDS:
-
acute respiratory distress syndrome
- AU:
-
arbitrary units
- DHR:
-
dihydrorhodamine
- LPS:
-
lipopolysaccharide
- MAS:
-
meconium aspiration syndrome
- MFI:
-
median fluorescence intensity
- PE:
-
phycoerythrin
- PMA:
-
phorbol-12-β-myristat-13-α-acetate
- TCC:
-
terminal SC5b-9 complement complex
References
Burgess AM, Hutchins GM 1996 Inflammation of the lungs, umbilical cord and placenta associated with meconium passage in utero. Review of 123 autopsied cases. Pathol Res Pract 192: 121–1128
Cleary GM, Wiswell TE 1998 Meconium-stained amniotic fluid and the meconium aspiration syndrome. An update. Pediatr Clin North Am 45: 11–529
Kattwinkel J 2002 Surfactant lavage for meconium aspiration: a word of caution. Pediatrics 109: 167–1168
Soukka H, Viinikka L, Kaapa P 1998 Involvement of thromboxane A2 and prostacyclin in the early pulmonary hypertension after porcine meconium aspiration. Pediatr Res 44: 38–842
Li YH, Yan ZQ, Brauner A, Tullus K 2001 Meconium induces expression of inducible NO synthase and activation of NF-κB in rat alveolar macrophages. Pediatr Res 49: 20–825
Khan AM, Elidemir O, Epstein CE, Lally KP, Xue H, Blackburn M, Larsen GL, Colasurdo GN 2002 Meconium aspiration produces airway hyperresponsiveness and eosinophilic inflammation in a murine model. Am J Physiol 283: 785–L790
Yigit S, Tekinalp G, Oran O, Yurdakok M, Aliefendioglu D, Gurgey A 2002 Endothelin 1 concentrations in infants with meconium stained amniotic fluid. Arch Dis Child Fetal Neonatal Ed 87: 212–F213
Kytola J, Kaapa P, Uotila P 2003 Meconium aspiration stimulates cyclooxygenase-2 and nitric oxide synthase-2 expression in rat lungs. Pediatr Res 53: 31–736
Moraes TJ, Chow CW, Downey GP 2003 Proteases and lung injury. Crit Care Med 31: 189–S194
Castellheim A, Lindenskov PH, Pharo A, Fung M, Saugstad OD, Mollnes TE 2004 Meconium is a potent activator of complement in human serum and in piglets. Pediatr Res 55: 10–318
Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, Videm V, Lappegard KT, Kohl J, Lambris JD 2002 Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 100: 869–1877
Fung M, Lu M, Fure H, Sun W, Sun C, Shi NY, Du Y, Su J, Swanson X, Mollnes TE 2003 Pre-neutralization of C5a-mediated effects by the monoclonal antibody 137-26 reacting with the C5a moiety of native C5 without preventing C5 cleavage. Clin Exp Immunol 133: 60–169
Fung M, Loubser PG, Undar A, Mueller M, Sun C, Sun WN, Vaughn WK, Fraser CD Jr 2001 Inhibition of complement, neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J Thorac Cardiovasc Surg 122: 13–122
Undar A, Eichstaedt HC, Clubb FJ Jr, Fung M, Lu M, Bigley JE, Vaughn WK, Fraser CD Jr 2002 Novel anti-factor D monoclonal antibody inhibits complement and leukocyte activation in a baboon model of cardiopulmonary bypass. Ann Thorac Surg 74: 55–362
Mollnes TE, Lea T, Frøland SS, Harboe M 1985 Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol 22: 97–202
Korhonen K, Soukka H, Halkola L, Peuravuori H, Aho H, Pulkki K, Kero P, Kaapa PO 2003 Meconium induces only localized inflammatory lung injury in piglets. Pediatr Res 54: 92–197
Guo RF, Ward PA 2002 Mediators and regulation of neutrophil accumulation in inflammatory responses in lung: insights from the IgG immune complex model. Free Radic Biol Med 33: 03–310
Rosenbloom AJ, Pinsky MR, Napolitano C, Nguyen TS, Levann D, Pencosky N, Dorrance A, Ray BK, Whiteside T 1999 Suppression of cytokine-mediated β2-integrin activation on circulating neutrophils in critically ill patients. J Leukoc Biol 66: 3–89
Hogg N, Leitinger B 2001 Shape and shift changes related to the function of leukocyte integrins LFA-1 and Mac-1. J Leukoc Biol 69: 93–898
Terregino CA, Lubkin CL, Thom SR 1997 Impaired neutrophil adherence as an early marker of systemic inflammatory response syndrome and severe sepsis. Ann Emerg Med 29: 00–403
Rainer TH 2002 L-selectin in health and disease. Resuscitation 52: 27–141
Heller AR, Groth G, Heller SC, Breitkreutz R, Nebe T, Quintel M, Koch T 2001 N-acetylcysteine reduces respiratory burst but augments neutrophil phagocytosis in intensive care unit patients. Crit Care Med 29: 72–276
Gerard C 2003 Complement C5a in the sepsis syndrome—too much of a good thing?. N Engl J Med 348: 67–169
Huber-Lang MS, Younkin EM, Sarma JV, McGuire SR, Lu KT, Guo RF, Padgaonkar VA, Curnutte JT, Erickson R, Ward PA 2002 Complement-induced impairment of innate immunity during sepsis. J Immunol 169: 223–3231
Zeiher BG, Matsuoka S, Kawabata K, Repine JE 2002 Neutrophil elastase and acute lung injury: prospects for sivelestat and other neutrophil elastase inhibitors as therapeutics. Crit Care Med 30: 281–S287
Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS 1999 Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 282: 4–61
Spragg RG 2002 The future of surfactant therapy for patients with acute lung injury—new requirements and new surfactants. Biol Neonate 81( suppl 1): 0–24
Cheng IW, Ware LB, Greene KE, Nuckton TJ, Eisner MD, Matthay MA 2003 Prognostic value of surfactant proteins A and D in patients with acute lung injury. Crit Care Med 31: 0–27
Terada LS 2002 Oxidative stress and endothelial activation. Crit Care Med 30: 186–S191
Abraham E 2003 Neutrophils and acute lung injury. Crit Care Med 31: 195–S199
Clark P, Duff P 1995 Inhibition of neutrophil oxidative burst and phagocytosis by meconium. Am J Obstet Gynecol 173: 301–1305
Kaapa P, Kytola J, Soukka H, Ahotupa M 1997 Human meconium has potent antioxidative properties. Biol Neonate 72: 1–75
Soukka HR, Ahotupa M, Ruutu M, Kaapa PO 2002 Meconium stimulates neutrophil oxidative burst. Am J Perinatol 19: 79–284
Wiswell TE, Fuloria M 1998 The meconium aspiration syndrome: the saga continues. Indian Pediatr 35: 059–1062
Fuloria M, Wiswell TE 1999 Resuscitation of the meconium-stained infant and prevention of meconium aspiration syndrome. J Perinatol 19: 34–241
Wiswell TE 2001 Handling the meconium-stained infant. Semin Neonatol 6: 25–231
Saugstad OD 2001 Resuscitation of newborn infants with room air or oxygen. Semin Neonatol 6: 33–239
Weinberger B, Weiss K, Heck DE, Laskin DL, Laskin JD 2001 Pharmacologic therapy of persistent pulmonary hypertension of the newborn. Pharmacol Ther 89: 7–79
Wiswell TE, Knight GR, Finer NN, Donn SM, Desai H, Walsh WF, Sekar KC, Bernstein G, Keszler M, Visser VE, Merritt TA, Mannino FL, Mastrioianni L, Marcy B, Revak SD, Tsai H, Cochrane CG 2002 A multicenter, randomized, controlled trial comparing Surfaxin (Lucinactant) lavage with standard care for treatment of meconium aspiration syndrome. Pediatrics 109: 081–1087
Glantz JC 2002 Clearing up meconium: clinical management and research ethics. Birth 29: 37–140
Liu WF, Harrington T 2002 Delivery room risk factors for meconium aspiration syndrome. Am J Perinatol 19: 67–378
Watford WT, Smithers MB, Frank MM, Wright JR 2002 Surfactant protein A enhances the phagocytosis of C1q-coated particles by alveolar macrophages. Am J Physiol 283: 1011–L1022
Laudes IJ, Chu JC, Sikranth S, Huber-Lang M, Guo RF, Riedemann N, Sarma JV, Schmaier AH, Ward PA 2002 Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am J Pathol 160: 867–1875
Bhole D, Stahl GL 2003 Therapeutic potential of targeting the complement cascade in critical care medicine. Crit Care Med 31: 97–S104
Hotchkiss RS, Karl IE 2003 The pathophysiology and treatment of sepsis. N Engl J Med 348: 38–150
Riedemann NC, Guo RF, Ward PA 2003 Novel strategies for the treatment of sepsis. Nat Med 9: 17–524
Guo RF, Riedemann NC, Ward PA 2004 Role of C5a-C5aR interaction in sepsis. Shock 21: 1–7
Koenig JM, Simon J, Anderson DC, Smith E, Smith CW 1996 Diminished soluble and total cellular L-selectin in cord blood is associated with its impaired shedding from activated neutrophils. Pediatr Res 39: 16–621
Kim SK, Keeney SE, Alpard SK, Schmalstieg FC 2003 Comparison of L-selectin and CD11b on neutrophils of adults and neonates during the first month of life. Pediatr Res 53: 32–136
Sprong T, Brandtzaeg P, Fung M, Pharo AM, Hoiby EA, Michaelsen TE, Aase A, van der Meer JW, van Deuren M, Mollnes TE 2003 Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood 102: 702–3710
Dobrovolskaia MA, Vogel SN 2002 Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect 4: 03–914
Swierzko AS, Cedzynski M, Kirikae T, Nakano M, Klink M, Kirikae F, Ziolkowski A, Vinogradov EV, Kawakami M 2003 Role of the complement-lectin pathway in anaphylactoid reaction induced with lipopolysaccharide in mice. Eur J Immunol 33: 842–2852
Riedemann NC, Guo RF, Hollmann TJ, Gao H, Neff TA, Reuben JS, Speyer CL, Sarma JV, Wetsel RA, Zetoune FS, Ward PA 2004 Regulatory role of C5a in LPS-induced IL-6 production by neutrophils during sepsis. FASEB J 18: 70–372
Sprong T, Moller AS, Kierulf P, Wedege E, Bjerre A, van der Meer JW, van Deuren M, Brandtzaeg P, Mollnes TE 2004 Complement activation and complement-dependent inflammation by Neisseria meningitidis are independent of lipopolysaccharide. Infect Immun 72: 344–3349
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was approved by the institutional review board and funded by the Research Council of Rikshospitalet, the Norwegian Foundation for Health and Rehabilitation, Tanox Inc., Houston, TX, and Gythfeldt's legacy.
Rights and permissions
About this article
Cite this article
Castellheim, A., Pharo, A., Fung, M. et al. Complement C5a Is a Key Mediator of Meconium-Induced Neutrophil Activation. Pediatr Res 57, 242–247 (2005). https://doi.org/10.1203/01.PDR.0000150725.78971.30
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000150725.78971.30
This article is cited by
-
The reduction rate of serum C3 following liver transplantation is an effective predictor of non-anastomotic strictures
Hepatology International (2014)
-
The role of complement in meconium aspiration syndrome
Journal of Perinatology (2008)
-
Role of complement C5a in mechanical inflammatory hypernociception: potential use of C5a receptor antagonists to control inflammatory pain
British Journal of Pharmacology (2008)
-
Dysfunction of innate immunity and associated pathology in neonates
The Indian Journal of Pediatrics (2007)
