
Abstract
X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets, hereditary hypophosphatemic rickets with hypercalciuria, and tumor-induced osteomalacia share clinical and biochemical features, and are collectively referred to as hypophosphatemic rickets (HR). Recently, the molecular bases of HR were elucidated. A review of medical records and mutational analyses of the PHEX and FGF23 genes were performed on 17 unrelated Korean children with HR. The male-to-female ratio was 3:14, and 5 patients were familial. Initial laboratory tests revealed typical features of HR. Seven different PHEX mutations were detected in 8 patients: 2 missense mutations, 2 nonsense mutations, and 3 short deletions. No functional FGF23 mutation was detected in any patient. Patients with the PHEX mutation tended to have more severe skeletal disease than those without. Of the patients with this mutation, no genotype-phenotype correlation and no gene dosage effect were noted. Treatment with vitamin D and phosphate resulted in only a partial growth improvement in most cases, and was frequently complicated by hypercalciuria, hypercalcemia, nephrocalcinosis, or hyperparathyroidism. Renal glycosuria was detected in six cases and was associated with more severe skeletal disease. We conclude that current HR treatment is not fully safe or effective, and that close monitoring of treatment effectiveness and for complications should be performed during long-term treatment. No genotype-phenotype correlation in XLH was detected in this study, but a large-scaled study on this topic is warranted. The large proportion of patients with a normal genetic study suggests the possibility of other causative gene(s).
Similar content being viewed by others
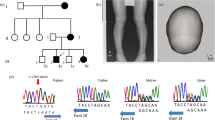

Main
HR is defined as a group of rachitic bone diseases associated with chronic hypophosphatemia, which results from defects in the renal tubular reabsorption of filtered phosphate (1–8). Clinically, XLH, ADHR, HHRH, and TIO can be categorized as HR (1–8).
XLH is the most common form of hereditary HR (2–6) and is caused by loss-of-function mutations in the PHEX gene (phosphate regulating gene with homologies to endopeptidases on the X chromosome). On the other hand, ADHR is far less common than XLH (2–6), and is associated with a gain-of-function mutation in the FGF23 gene encoding FGF23. The mutant FGF23 molecule is resistant inactivation by proteolytic cleavage (2,5–12). Moreover, TIO is an acquired form of HR, which is caused by a variety of benign primitive mesenchymal tumors that secrete FGF23 (2,7,13). HHRH is a rare hereditary disease and its underlying pathogenesis remains to be elucidated.
Several studies of the molecular defects in XLH, ADHR, and TIO support a model of a common pathogenetic mechanism in these three diseases (2,6,14). In this model, FGF23 has been suggested to be the main circulating phosphaturic factor (2,6,14), and the circulating level of FGF23 is determined in part by the rate of its proteolytic cleavage by PHEX protease (9,13,15). The common abnormality shared by loss-of-function mutations in the PHEX gene (as in XLH), gain-of-function mutations in the FGF23 gene (as in ADHR), and overproduction of FGF23 by tumors (as in TIO) is an elevation of circulating FGF23 (2,5–8,13,14). However, current studies (16,17) fail to confirm PHEX-dependent cleavage of FGF23 and suggest that the increased circulating level of FGF23 in XLH reflects, at least in part, an increase in its production (17). These findings support a more complex model of pathogenesis that includes intermediate steps linking PHEX and FGF23. One possibility is that increased FGF23 expression results from the actions of yet to be identified PHEX substrate or from other downstream consequences of loss-of-function mutations in PHEX (17). The increased circulating level of active FGF23 suppresses Na-phosphate cotransport activities both in kidney and in small intestine, as well as 25(OH)D3 1α-hydroxylase mRNA expression in the kidney (13,18). The final phenotype is rachitic bone changes associated with chronic hypophosphatemia and an inappropriately low serum level of 1α,25-dihydroxy vitamin D3.
In the present study, a clinical and molecular genetic analysis was performed in children with a clinical diagnosis of HR.
METHODS
Seventeen unrelated Korean children, diagnosed with HR by the Department of Pediatrics, Seoul National University Children's Hospital, were enrolled in this study. One patient who had hypercalciuria before medication was excluded, and no case was diagnosed as TIO. The medical records of these cases were reviewed retrospectively. For phenotypic analysis, the severities of the two principal disease manifestations, i.e. skeletal or dental diseases were determined (3,19) and initial laboratory data were obtained. Skeletal disease was graded as “mild” or “severe” based on the need for orthopedic surgery. Hypercalciuria was defined as a urine calcium to creatinine ratio >0.25, and hypercalcemia as a serum total calcium level of >11 mg/dL. The presence of nephrocalcinosis was determined by increased renal medullary echogenicity in renal ultrasonography. Hyperparathyroidism was defined by an increasing serum intact PTH (iPTH) >150 pg/mL (normal, 10–65 pg/mL). To evaluate the effect of therapy, SDS of height was calculated in 13 patients who had been treated for at least 2 y.
Mutational analyses of the PHEX and FGF23 genes were carried out in all 17 patients and in available family members. For the PHEX gene, total peripheral blood mRNA were reverse transcribed to cDNA, and three overlapping cDNA fragments covering the entire coding sequences of the PHEX gene were amplified by nested PCR using primers listed in Table 1 and directly sequenced. The three exons of the FGF23 gene were amplified from peripheral blood genomic DNA by PCR and directly sequenced.
To evaluate the significance of the T239M mutation in the FGF23 gene, which was detected in some of the patients, the presence of the T239M genotype was determined in 50 healthy controls. These studies were approval by the Ethics Committee of Seoul National University Hospital, Seoul, Korea, and informed consent for the genetic analysis was obtained from all patients or their parents and healthy controls.
Statistical analysis was conducted using the two-tailed Fisher exact test, the Mann-Whitney U test, the paired t test, and the Wilcoxon rank-sum test. A p value > 0.05 was considered statistically insignificant.
RESULTS
The male-to-female ratio of the 17 patients was 3:14. Median ages at onset and diagnosis were 20 mo (10–35 mo) and 35 mo (10–60 mo), respectively. Five cases (29%) were familial, and the modes of inheritance were dominant (one parent was affected at least, and there was no father-to-son transmission). The presenting manifestations include bowlegs (100%), gait disturbance (18%), and lordosis (6%). Dental problems, such as abscess or caries, were detected initially or developed during follow-up in six cases (35%). Initial laboratory findings before treatment were as follows: mean total calcium and phosphorus serum levels were 9.5 ± 0.7 and 2.5 ± 0.4 mg/dL, respectively, and the mean alkaline phosphatase level was 727 ± 207 IU/L. The serum 25(OH)D3 and 1,25(OH)2D3 levels were 28 ± 14 ng/mL (normal, 10–55 ng/mL) and 44 ± 20 pg/mL (normal, 20–76 pg/mL), respectively. The serum iPTH was 43 ± 21 pg/mL (normal, 16–65 pg/mL), and osteocalcin was 53 ± 57 ng/mL (normal, 1.8–6.6 ng/mL). Tubular reabsorption of phosphate and transport maximum of phosphate were 48.9 ± 22.6% (normal, 83–93%) and 1.33 ± 0.56 mg/dL of GFR (normal, 3.7–5.3 mg/dL of GFR), respectively. The random urine calcium to creatinine ratio was 0.04 ± 0.03.
PHEX gene analysis revealed seven different mutations in eight patients (47%), including two missense mutations, two nonsense mutations, and three short deletions (Table 2). The R20X mutation was detected in two unrelated patients. All missense and nonsense mutations detected had already been reported, and three short deletions were novel. Among the eight patients with a PHEX mutation, three cases (38%) were familial and five (63%) were sporadic, both genetically and clinically. Nine patients with no PHEX mutation were female, and two of these had a positive family history (father and two daughters in one family, and mother and a daughter in another family). In three patients with no PHEX mutation and no family history, a heterozygous 239Thr(ACG)>Met(ATG) change in the FGF23 gene was detected. The distribution of genotypes for this T239M in 50 healthy controls was as follow: 239 TT in 34 (68%) and 239 TM in 16 (32%).
The clinical and laboratory findings were compared between patients with and without PHEX mutations. All patients without a PHEX mutation were female. Skeletal disease appeared more severe with higher serum alkaline phosphatase levels in patients with the PHEX mutation, but the statistical significance was only marginal (p = 0.059) (Table 3). Genotype-phenotype correlations were analyzed in eight patients with the PHEX mutation, but no significant correlation was found between the severity of skeletal or dental diseases and mutation types (Table 4). In addition, the effect of gender on phenotype and the gene dosage effect were analyzed in eight patients with the PHEX mutation, but no significant relation was found (Table 5).
All patients were treated with a combination of oral phosphate supplementation and vitamin D (1,25(OH)2D3 or 1-α(OH)D3). Orthopedic surgery, such as corrective osteotomy or epiphysiodesis, was administered in three patients (18%), and a brace was applied in 12 (71%). The SDS of height of 13 patients before and after treatment for 2 y revealed an improvement from −2.23 ± 1.00 to −1.70 ± 1.09 (p < 0.05). However, complete catch-up growth was not achieved in most cases (Fig. 1). During follow-up, several complications associated with medications were detected as follows: hypercalciuria in 14 cases (82%), hypercalcemia in 7 (41%), nephrocalcinosis in 10 (59%), and hyperparathyroidism in 4 (24%). All of these complications, except nephrocalcinosis, were intermittent or transient. In addition, microscopic hematuria and renal glycosuria were found in 4 (24%) and 6 cases (35%), respectively. Microscopic hematuria was detected with concomitant hypercalciuria in all cases. Patients without renal glycosuria had milder skeletal disease than those with renal glycosuria (p = 0.03) (Table 6). Renal function was maintained normally in all cases.

SDS of height (Ht SDS) calculated in 13 patients before and after treatment for 2 y. The Ht SDS was improved in nine patients after treatment. However, complete catch-up growth was achieved in only one of them.
DISCUSSION
XLH, which is caused by loss-of-function mutations in the PHEX gene, is the most common form of hereditary HR (2–6). In an online PHEX mutation database (http://www.phexdb.mcgill.ca), a total of 176 mutations in PHEX were listed in June 2004. Mutation types are variable and include frame shifts, missense mutations, abnormal splicings, nonsense mutations, and deletions. The mutational detection rates in XLH patients in most reports are approximately 65%, but are significantly lower (23–47%) in sporadic cases without family history as 23–47% (3,20), except for one Finnish report (21), which found detection rates in familial and sporadic cases of 100% and 93%, respectively. The detection rate found in the present study was lower (47%) than in other reports. However, when familial and sporadic cases were separated, mutations were detected in 60% of the former and 42% of the latter, which concurs with other reports. At this moment, it is difficult to confirm whether the larger proportion of sporadic cases (71%) in the present study compared with other reports means a higher incidence of de novo occurrence in Koreans. In general, about one-third of XLH patients showed no mutation in the coding region of the PHEX gene. And, it remains to be determined whether they harbor mutations in the promoter, 3′ untranslated region, or intronic regions of the gene. Christie et al. (22) found pseudoexons of the gene in some patients. In the present study, intronic mutations or abnormal splicings including pseudoexons should have detected if present, because RT-PCR was used. However, because nine patients with no PHEX mutation in the present study were female, another technical problem may have prevented the detection of heterozygous mutations. If a mutation occurred within a very early fragment of the gene, a mature transcript may not have been made. In this instance, examining the DNA encoding individual exons gives a higher chance for finding a mutation, especially in female XLH patients where both PHEX alleles are transcribed. The possibility of another form of HR with an extreme female preponderance associated with an unknown gene should also be considered. We found a heterozygous T239M change in the FGF23 gene in 3 (33%) of 9 patients without PHEX mutations and in 16 (32%) of 50 healthy subjects. The ADHR Consortium (12) had previously reported T239M as a polymorphism with similar allelic frequencies (threonine in 89.7%, methionine in 10.3%).
The phenotype-genotype correlation in XLH patients has been studied in few papers (3,23). In one of those studies, the clinical symptoms did not strictly depend upon the type and localization of the PHEX gene mutation (23). However, hearing defect was correlated with mutations in the beginning fragment, and dental disease and increased head length with the mutations in the beginning and the terminal fragment of the gene. In another study of 50 patients, including 31 with a PHEX mutation (3), no correlation was found between disease severity and the type or location of PHEX mutation. And no difference was found in the severity of skeletal and dental disease between hemizygous males and heterozygous females. Therefore, the identification of PHEX mutations in XLH patients may be of limited prognostic value (3). Although the number of patients in the present study was not large enough to obtain a statistically meaningful value, we obtained the same negative results. As an X-linked disorder, the absence of a gene dosage effect in XLH is unusual. This can be proven by comparing the phenotypes of male and female patients for common mutations such as R20X.
Although renal glycosuria is one of the known manifestations of XLH, its underlying pathophysiologic mechanism has not been fully elucidated. It is suggested that disease-causing circulating phosphaturic factors in XLH patients inhibit the Na-phosphate transporter and Na-glucose transporter in proximal renal tubular cells (24–26). Inappropriate glycosuria after a glucose load was observed in all Hyp mice, an animal model, whereas it only occurred in a fraction of XLH patients (24,25). This difference may reflect gene penetrance differences. In addition, renal glycosuria after glucose ingestion accompanied by a reduction in renal phosphate reabsorption, and baseline renal phosphate reabsorption was lower in patients who developed glycosuria (24–26). These findings suggest that excessive glucose ingestion may reduce the efficacy of oral phosphate therapy in XLH patients, and that patients with renal glycosuria may have a more severe phenotype. In the present study, although baseline renal phosphate reabsorption was not significantly different between patients who developed glycosuria and those who did not, skeletal disease was more severe in the former group of patients.
Current treatment for XLH consists of high-dose oral phosphate supplementation in combination with calcitriol, which should be continued at least until growth is complete. However, this form of therapy is far from being safe and effective (2,27). Although the correction of rickets and growth improvement can be expected in most patients to some degree on this treatment, complete growth catch-up is not achieved in most cases and long-term treatment is frequently complicated by hyperparathyroidism, hypercalcemia, hypercalciuria, or nephrocalcinosis, as shown in this study (28–30). In addition, long-term compliance is difficult in most patients. Thus, novel therapeutic strategies, based on new knowledge of the molecular pathogenesis of HR, are clearly required.
Abbreviations
- ADHR:
-
autosomal dominant hypophosphatemic rickets
- FGF23:
-
fibroblast growth factor 23
- HHRH:
-
hereditary hypophosphatemic rickets with hypercalciuria
- HR:
-
hypophosphatemic rickets
- SDS:
-
standard deviation score
- TIO:
-
tumor-induced osteomalacia
- XLH:
-
X-linked hypophosphatemic rickets
References
Hanna JD, Niimi K, Chan JC 1991 X-linked hypophosphatemia. Genetic and clinical correlates. Am J Dis Child 145: 865–870
Tenenhouse HS, Murer H 2003 Disorders of renal tubular phosphate transport. J Am Soc Nephrol 14: 240–248
Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, Cowell CT, Carpenter TO 2001 Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 86: 3889–3899
Drezner MK 2000 PHEX gene and hypophosphatemia. Kidney Int 57: 9–18
Rowe PS 2000 The molecular background to hypophosphatemic rickets. Arch Dis Child 83: 192–194
Quarles LD 2003 FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab 285: E1–E9
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanish Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H 2003 Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348: 1656–1663
Strewler GJ 2001 FGF23, hypophosphatemia, and rickets: has phosphatonin been found?. Proc Natl Acad Sci U S A 98: 5945–5946
Chan JC, Alon U, Hirschman GM 1985 Renal hypophosphatemic rickets. J Pediatr 106: 533–544
Jan de Beur SM, Levine MA 2002 Molecular pathogenesis of hypophosphatemic rickets. J Clin Endocrinol Metab 87: 2467–2473
Bai XY, Miao D, Goltzman D, Karaplis AC 2003 The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 278: 9843–9849
White KE, Evans WE, O'Riordan JL, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM, the ADHR Consortium 2000 Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23.. Nat Genet 26: 345–348
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T 2001 Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A 98: 6500–6505
Strom TM, Francis F, Lorenz B, Boddrich A, Econs MJ, Lehrach H, Meitinger T 1997 Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet 6: 165–171
Sabbagh Y, Boileau G, DesGroseillers L, Tenenhouse HS 2001 Disease-causing missense mutations in the PHEX gene interfere with membrane targeting of the recombinant protein. Hum Mol Genet 10: 1539–1546
Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD 2003 Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 278: 37419–37426
Benet-Pagès A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM 2004 FGF23 is processed by proprotein convertases but not by PHEX. Bone 35: 455–462
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto KI, Fukushima N 2003 Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1α,25-dihydroxyvitamin D3 production. J Biol Chem 278: 2206–2211
Scriver CR, Tenenhouse HS 1992 X-linked hypophosphataemia: a homologous phenotype in humans and mice with unusual organ-specific gene dosage. J Inherit Metab Dis 15: 610–624
Dixon PH, Christie PT, Wooding C, Trump D, Grieff M, Holm I, Gertner JM, Schmidtke J, Shah B, Shaw N, Smith C, Tau C, Schlessinger D, Whyte MP, Thakker RV 1998 Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab 83: 3615–3623
Tyynismaa H, Kaitila I, Näntö-Salonen K, Ala-Houhala M, Alitalo T 2000 Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets. Hum Mutat 15: 383–384
Christie PT, Harding B, Nesbit MA, Whyte MP, Thakker RV 2001 X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. J Clin Endocrinol Metab 86: 3840–3844
Popowska E, Pronicka E, Sulek A, Jurkiewicz D, Rowinska E, Sykut-Cegielska J, Rump Z, Arasimowicz E, Krajewska-Walasek M 2001 X-linked hypophosphatemia in Polish patients. 2. Analysis of clinical features and genotype-phenotype correlation. J Appl Genet 42: 73–88
Muhlbauer RC, Fleisch H 1991 Abnormal renal glucose handling in X-linked hypophosphataemic mice. Clin Sci (Lond) 80: 71–76
Baran DT, Hahn TJ, Avioli LV 1978 Effect of oral glucose ingestion on renal phosphate reabsorption and clearance in vitamin D-resistant rickets. J Clin Endocrinol Metab 47: 138–144
Aubert J, Templier B, Lacour B, Clavel J, Ulmann A 1984 Modification in the renal phosphate handling after oral administration of glucose to conscious rats. Miner Electrolyte Metab 10: 98–102
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M 1991 Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med 325: 1843–1848
Mughal Z 2002 Rickets in childhood. Semin Musculoskelet Radiol 6: 183–190
Makitie O, Kooh SW, Sochett E 2003 Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol (Oxf) 58: 163–168
Alon US, Monzavi R, Lilien M, Rasoulpour M, Geffner ME, Yadin O 2003 Hypertension in hypophosphatemic rickets-role of secondary hyperparathyroidism. Pediatr Nephrol 18: 155–158
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (02-PJ1-PG3–21001–0012).
Rights and permissions
About this article
Cite this article
Cho, H., Lee, B., Kang, J. et al. A Clinical and Molecular Genetic Study of Hypophosphatemic Rickets in Children. Pediatr Res 58, 329–333 (2005). https://doi.org/10.1203/01.PDR.0000169983.40758.7B
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000169983.40758.7B
This article is cited by
-
The genetic polymorphisms of XPR1 and SCL34A3 are associated with Fanconi syndrome in Chinese patients of tumor-induced osteomalacia
Journal of Endocrinological Investigation (2021)
-
Phenotypic characterization of X-linked hypophosphatemia in pediatric Spanish population
Orphanet Journal of Rare Diseases (2021)
-
Novel variants and uncommon cases among southern Chinese children with X-linked hypophosphatemia
Journal of Endocrinological Investigation (2020)
-
Regulation of Fibroblast Growth Factor 23 by Iron, EPO, and HIF
Current Molecular Biology Reports (2019)
-
Hypophosphatemic rickets: etiology, clinical features and treatment
European Journal of Orthopaedic Surgery & Traumatology (2015)
