
Abstract
The peroxisome biogenesis disorders (PBDs) form a genetically and clinically heterogeneous group of disorders due to defects in at least 11 distinct genes. The prototype of this group of disorders is Zellweger syndrome (ZS) with neonatal adrenoleukodystrophy (NALD) and infantile Refsum disease (IRD) as milder variants. Common to PBDs are liver disease, variable neurodevelopmental delay, retinopathy and perceptive deafness. PBD patients belonging to complementation group 10 (CG10) have mutations in the PEX2 gene (PXMP3), which codes for a protein (PEX2) that contains two transmembrane domains and a zinc-binding domain considered to be important for its interaction with other proteins of the peroxisomal protein import machinery. We report on the identification of four PBD patients belonging to CG10. Sequence analysis of their PEX2 genes revealed 4 different mutations, 3 of which have not been reported before. Two of the patients had homozygous mutations leading to truncated proteins lacking both transmembrane domains and the zinc-binding domain. These mutations correlated well with their severe phenotypes. The third patient had a homozygous mutation leading to the absence of the zinc-binding domain (W223X) and the fourth patient had a homozygous mutation leading to the change of the second cysteine residue of the zinc-binding domain (C247R). Surprisingly, the patient lacking the domain had a mild phenotype, whereas the C247R patient had a severe phenotype. This might be due to an increased instability of PEX2 due to the R for C substitution or to a dominant negative effect on interacting proteins.
Similar content being viewed by others

Main
The peroxisome biogenesis disorders (PBDs; MIM #601539), which comprise Zellweger syndrome (ZS; MIM #214100), neonatal adrenoleukodystrophy (NALD; MIM #202370) and infantile Refsum disease (IRD; MIM #266510), represent a spectrum of disease severity with ZS being the most, and IRD the least severe disorder. Common to all three PBDs are liver disease, variable neurodevelopmental delay, retinopathy and perceptive deafness (1). Patients with ZS are severely hypotonic from birth and die before 1 year of age. Patients with NALD experience neonatal onset of hypotonia and seizures and suffer from progressive white matter disease, dying usually in late infancy (2). Patients with IRD may survive beyond infancy and some may even reach adulthood (3). Clinical differentiation between these disease states is not very well-defined and patients can have overlapping symptoms (4).
The absence of functional peroxisomes in patients with a PBD leads to a number of biochemical abnormalities. PBD patients have an impaired synthesis of plasmalogens, due to a deficiency of the two enzymes dihydroxyacetonephosphate acyltransferase (DHAPAT) and alkyl-dihydroxyacetonephosphate synthase (5, 6). Peroxisomal fatty acid β-oxidation is also defective, leading to the accumulation of very-long chain fatty acids (VLCFAs), notably C26:0, the branched chain fatty acid pristanic acid and the bile acid intermediates di- and trihydroxycholestanoic acid (DHCA and THCA) (1). Phytanic acid α-oxidation and L-pipecolic acid oxidation are also impaired (1). In contrast, some peroxisomal enzymes show normal activity including catalase, D-amino acid oxidase, L-α-hydroxy acid oxidase A and alanine:glyoxylate aminotransferase, although subcellular fractionation studies have shown that these enzymes are mislocalized in the cytoplasm (1).
The PBDs are caused by genetic defects in PEX genes encoding proteins called peroxins, which are required for the biogenesis of peroxisomes and function in the assembly of the peroxisomal membrane or in the import of enzymes into the peroxisome (7). After synthesis on free polyribosomes, peroxisomal matrix proteins carrying either a carboxy-terminal peroxisomal targeting sequence 1 (PTS1) or a cleavable amino-terminal PTS2 signal are translocated across the peroxisomal membrane (8, 9). A defect in one of the peroxins of the peroxisomal import machinery leads to failure of protein import via the PTS1- and/or PTS2-dependent import pathway, and consequently to functional peroxisome deficiency. Cell fusion complementation studies using patient fibroblasts revealed the existence of at least 11 distinct genetic groups of which currently all corresponding PEX genes have been identified. Most complementation groups are associated with more than one clinical phenotype (7).
PBD patients belonging to CG10 (CG F according to the Japanese nomenclature) have mutations in the PEX2 gene (PXMP3: MIM #170993) (10). The PEX2 gene was the first gene found to be mutated in ZS and spans approximately 17.5kb in length and contains four exons. The entire coding sequence is included in exon 4 (11). The gene encodes a 305 amino acid protein (PEX2), with a molecular weight of ∼35 kD. PEX2 is an integral membrane protein with two transmembrane domains, exposing its NH2 and COOH termini to the cytoplasm (12). PEX2 contains a zinc-binding motif (C3HC4) at the C-terminal part, probably involved in interaction with the other proteins of the peroxisomal protein import machinery. PEX10 and PEX12 also contain similar zinc-binding motifs. PEX2 was shown to interact with PEX10 (13), and was present in a complex consisting of PEX2, PEX5, PEX12, and PEX14 (14).
In this study we report the identification of novel mutations in the PEX2 gene in four PBD patients which, using cell fusion complementation analysis, were shown to belong to complementation group 10. The correlation between genotypes and phenotypes is discussed.
METHODS
Subjects.
All patients analyzed showed the clinical characteristics of PBDs. After we obtained informed consent, samples were collected from patients and sent to our laboratory for biochemical and molecular diagnosis. The biochemical diagnosis of a PBD was substantiated by detailed studies in primary skin fibroblasts, which included the following analyses:de novo plasmalogen synthesis, DHAPAT activity, C26:0 and pristanic acid β-oxidation, VLCFA levels, phytanic acid α-oxidation, catalase immunofluorescence and immunoblot analysis of peroxisomal thiolase and acyl-CoA oxidase (15).
Case reports.
Patient 1 was a male infant, first child of consanguineous Moroccan parents, born after an uneventful pregnancy with low birth weight (2290 g) for gestational age (41 wk). He was severely hypotonic with absent tendon reflexes and had a large anterior fontanelle and metopic sutures, a high forehead, slight hepatomegaly, cryptorchidism, hypospadias and a cardiac murmur on auscultation. He was transferred to the neonatal intensive care unit because of generalized convulsions and myoclonic jerks. Neuroimaging of the brain (MRI) showed a complete absence of the corpus callosum, colpocephaly, pachygyria, leucomalacia, and subcortical and periventricular and cerebellar hypoplasia. EEG abnormalities were not specific and showed diffuse epileptic activity. Ocular abnormalities included a pendular nystagmus, cataracts, optic atrophy and a negative visual evoked response (VER). There was an impaired hearing with reduced brainstem auditory evoked potentials (BAEP). There were no skeletal abnormalities. Ultrasonography of the kidneys showed no abnormalities. Cardiac defects included insufficiency of the mitral, tricuspid and aortic valves and a peripheral pulmonary artery stenosis. From the 1st day he developed a severe icterus with elevated serum liver enzymes (ASAT, ALAT, LDH) and a predominance of serum conjugated bilirubine (cholestasis). A liver biopsy showed severe cholestasis with mitochondrial abnormalities (absence of cristae) on electron microscopy (EM) and absence of peroxisomes and catalase activity present in the cytoplasm of the hepatocytes revealed by immunohistochemical examinations. A skin biopsy showed spicular inclusions in a Schwan cell on EM as has been described in adrenomyeloneuropathy. Biochemical abnormalities included high serum levels of VLCFA, very low plasmalogen content of red blood cell membranes and the presence of THCA and C29 dicarboxylic acid in urine. The course of the disease was rapid: difficulties with sucking and swallowing necessitated gavage feeding; convulsions persisted under therapy with phenobarbital and vigabatrin; the cholestatic icterus worsened with age and his general condition deteriorated progressively. He died at the age of 3 mo.
Patient 2 has been described before as the third child of Israeli Arab, 1st degree cousins (16). He was born at term, after an uncomplicated pregnancy and delivery. On routine examination at 9 mo of age he was reported as an alert, well developed baby without dysmorphic features, hepatomegaly or neurologic abnormalities. At age 22 mo he could not walk unassisted, had hypotonia with absent tendon reflexes and athetoid movements. MRI revealed cerebellar and vermian atrophy and dysmyelination. At age 4 y he had retinitis pigmentosa, a flattened electroretinogram, abnormal visual evoked potentials and sensorineural hearing loss documented by abnormal BAEP. Motor and sensory nerve conductions were prolonged. Results of screening of urine and plasma for abnormal amino acids, organic acids, oligosacharides and purine and pyrimidine metabolites were negative. Lysosomal enzyme activities in fibroblasts were normal. The patient continued to deteriorate and was in a vegetative state at the age of 9 y. At this age he had elevated plasma levels of VLCFA, pipecolic and phytanic acid and abnormal bile acid intermediates which suggested a peroxisomal biogenesis defect. EM and immunocytochemical studies of the liver disclosed absence of peroxisomes in approximately 90% of hepatocytes. The remaining 10% of the hepatocytes however, had numerous normal looking peroxisomes containing catalase, alanine-glyoxylate aminotransferase and peroxisomal β-oxidation enzymes. At that time, studies performed in cultured fibroblasts revealed normal β-oxidation of VLCFA and normal DHAPAT activity and a normal catalase latency test (16). The child's condition continued to deteriorate and he died from pneumonia at age 13 y.
Patient 3 was a female infant, first child of nonconsanguineous parents from Ashkenazi-Jewish origin. At birth, she looked dysmorphic with epicanthal folds, broad nasal bridge, high palate, dysplastic ears, excessive skin on the upper back and neck, hypoplastic nipples, hypoplastic external genitalia and hypoplastic nails. She had a broad anterior fontanelle and her liver was enlarged. Her muscle tone was markedly decreased and there was paucity of spontaneous movements. Biochemical investigations in plasma revealed clear abnormalities indicative of a PBD as confirmed in fibroblasts in which a generalized loss of peroxisomal functions and the absence of peroxisomes were found. The patient died in early infancy. The parents had a second child, who was healthy. Their third child was a female, born at 42 wk. Her physical examination revealed dysmorphism similar to that of her older sister. Echocardiography revealed an atrial septal defect, a ventricular septal defect and tricuspid regurgitation. She was severely hypotonic and unable to suck. Seizures started on the 2nd day of life and continued till her death at 1 month.
Patient 4 was a full term male of first cousins. He had three healthy sisters. He was delivered after a normal pregnancy by an emergency perpartum caesarian section because of sudden fetal distress. He required immediate mechanical ventilation for the absence of respiratory movements. Generalized seizures were noted soon after birth and treated by i.v. diazepam. The patient was transferred to the intensive care unit. Lethargy, severe hypotonia, poor spontaneous movements and absence of sucking were noted, as well as an unusual large anterior fontanelle. The baby was not dysmorphic. Cardiac and respiratory examination were normal, hemodynamic status was stable and the liver was not enlarged. Initial laboratory investigations were normal. Because of the isolation of Staphylococcus aureus from the mother, neonatal infection was first considered. However, the patient's neurologic status did not improve and the seizures persisted despite the administration of antibiotics and anticonvulsants and the absence of meningitis. Brain MRI examination was normal, excluding severe perinatal asphyxia. Laboratory investigations at 7 wk of life evidenced moderate cytolysis (ASAT: 246 U/L, ALAT: 88 U/L, γGT: 387 U/L, alkaline phosphatase: 780 U/L). Liver ultrasound examination was normal but renal ultrasound showed two polycystic kidneys. X-ray skeleton was normal. Indirect ophthalmoscopy did not reveal any lesion. In contrast, signal on electroretinogram and visual evoked potentials were absent. Plasma VLCFAs were abnormally elevated (C24/C22 ratio: 1.76 (N: 0.86 ± 0.07) and C26/C22 ratio: 0.472 (N: 0.026 ± 0.016)). Pipecolic acid as well as C27 bile acid intermediates were elevated (pipecolic acid: 13.5 μmol/L (N: 0.54–2.46); THCA: 47.5 μmol/L (N < 0.035); DHCA: 61.9 μmol/L (N < 0.119)). These data were indicative of a PBD as confirmed in fibroblasts in which a generalized loss of peroxisomal functions and the absence of peroxisomes were found. Neurologic status progressively worsened and the patient died at the age of 2 mo.
Biochemical analysis.
DHAPAT activity (17), C26:0 and pristanic acid β-oxidation (18) were assayed in primary skin fibroblasts as previously described. Catalase immunofluorescence (19) and complementation analysis (20) were performed as described before. To allow complementation analysis in the cells of patient 2, we cultured these at 40°C for 3 d after fusion of the cells. This treatment results in a significant decrease of catalase-positive particles due to the mosaicism. Details on this method will be described elsewhere (Gootjes et al. in preparation).
Mutation analysis.
PEX2 mutation analysis in the patients was performed at the genomic DNA level. Genomic DNA was isolated from primary skin fibroblasts using the Wizard genomic DNA purification kit (Promega, Madison, WI). The entire exons plus flanking intron sequences from the PEX2 gene were amplified by PCR using the primer sets shown in Table 1. All forward and reverse primers used for mutation analysis were tagged with a -21M13 (5′-TGTAAAACGACGGCCAGT-3′) sequence and M13rev (5′-CAGGAAACAGCTATGACC-3′) sequence, respectively. PCR fragments were sequenced in two directions using ′-21M13′ and ′M13rev′ fluorescent primers on an Applied Biosystems 277A automated DNA sequencer following the manufacturer's protocol (Perkin Elmer, Wellesley, MA, U.S.A.).
Quantitative real-time RT-PCR analysis.
Total RNA was isolated from primary skin fibroblasts using Trizol (Invitrogen, Carlsbad, CA, U.S.A.) extraction, after which cDNA was prepared using a first strand cDNA synthesis kit for RT-PCR (Roche, Mannheim, Germany). Quantitative real-time PCR analysis of PEX2 and β- 2-microglobulin RNA was performed using the LightCycler FastStart DNA Master SYBR green I kit (Roche, Mannheim, Germany). PEX2 primers used were: PEX2-LC-F, 5′- GTCTCTGAGCTTCTGGCAAG -3′ and PEX2-LC-R, 5′-AAACTGGGACCAAACTAGCTG-3′. β-2-Microglobulin primers used were: b2M-FW, 5′-TGAATTGCTATGTGTCTGGG-3′ and b2M-REV, 5′-CATGTCTCGATCCCACTTAAC-3′. The PCR program comprised a 10 min initial denaturation step at 95°C to activate the hot start polymerase, followed by 40 cycles of 95°C for 10 s, 58°C for 2 s and 72°C for 11 s (9 s for β-2-microglobulin). Fluorescence was measured at 82°C for PEX2 and 80°C for β-2-microglobulin. Melt curve analysis to show generation of a single product for each reaction was carried out following the PCR program. Amplification of a single product of the correct size was also confirmed by agarose gel electrophoresis. Duplicate analysis was performed for all samples. Data were analyzed using LightCycler Software, version 3.5 (Roche, Mannheim, Germany). To adjust for variations in the amount of input RNA, the values for the PEX2 gene were normalized against the values for the housekeeping gene β-2-microglobulin and the patient ratios were presented as a percentage of the mean of 2 control fibroblast cell lines.
RESULTS
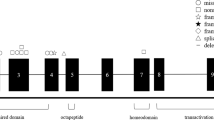
In this study we analyzed four patients affected by a PBD as concluded from the finding of typical abnormalities in plasma (elevated levels of VLCFA, bile acid intermediates, pristanic and phytanic acid) and primary skin fibroblasts (deficient DHAPAT activity, C26:0 β-oxidation and pristanic acid β-oxidation (Table 2)). Catalase immunofluorescence revealed the absence of peroxisomes in patient 1, 3 and 4, whereas patient 2 displayed a mosaic pattern, characterized by the absence of punctate immunofluorescence in the majority of cells whereas in about 30% of cells a punctate staining pattern was found. Subsequent cell fusion complementation studies revealed that the four patients belong to CG10 with PEX2 as the causative gene. Sequence analysis of the PEX2 gene of these patients revealed 4 different mutations, 3 of which have not been reported before. The mutations involve one missense mutation, two nonsense mutations and one deletion (Table 2). Patient 1 was homozygous for a missense mutation changing the cysteine at position 247 to an arginine (Fig. 1). This cysteine residue is the second cysteine residue of the zinc-binding domain. Patient 2 was homozygous for a nonsense mutation (W223X) that truncates the protein between the second transmembrane domain and the zinc-binding domain. Patient 3 was homozygous for a nonsense mutation (R119X) that truncates the protein before the first transmembrane domain. This mutation has been described before (10, 21). Patient 4 was homozygous for a 5-bp deletion (279-283delGAGAT) that results in a frameshift and leads to truncation of the protein before the first transmembrane domain. Thus, three of the four mutations create an early termination codon in the PEX2 open reading frame that will result in a truncated protein product (Fig. 2).

Deduced PEX2 products of 4 PBD patients. The diagram shows the predicted protein product of each PEX2 allele. The zinc-binding domain is indicated by a dark gray box and each of the transmembrane domains is indicated by the black boxes. The light gray bar indicates the length of additional amino acids that are appended as a result of a frameshifting mutation.

Quantitative real-time RT-PCR analysis of PEX2. Presented are the PEX2/β-2-microglobulin ratios expressed as percentages of the mean of controls 1 and 2.
In eukaryotic cells, the introduction of a nonsense codon into mRNA can also lead to nonsense-mediated decay of the mRNA, resulting in a reduction of protein production, a process common in human genetic disease (22, 23). To test for this latter possibility as a primary cause of PEX2 dysfunction in these patients, RNA from the patient cell lines was analyzed by real-time RT-PCR to quantify PEX2 mRNA. These analyses showed that PEX2 transcript levels in patient 1, carrying the missense mutation C247R, were elevated to 150%, compared with controls (Fig. 2). Of the three patients with frameshift or nonsense mutations, only patient 2 showed decreased PEX2 mRNA levels of around 35%. The PEX2 transcript levels in patient 3 and 4 were relatively normal with 90% and 80%, respectively.
DISCUSSION
Mutations in PEX2 are known to underlie the disease in patients with a PBD belonging to complementation group 10 (10). In this study we determined the PEX2 genotypes of four patients diagnosed in our laboratory. All four patients have mutations in the PEX2 gene, confirming that a defective PEX2 is responsible for the disease in these patients. The mutations involve one missense mutation, two nonsense mutations and one deletion. Three of the mutations have not been described previously. Except for patient 2, all patients had the severe ZS phenotype and died before 3 mo of age. Patient 2 was diagnosed with IRD and survived for over 9 y. For patient 3 and 4 the phenotype appears to correlate rather well with the genotype. The two mutations in both patients involve a stop codon upstream of the sequences encoding the transmembrane domains and result into a severe ZS phenotype. This strongly suggests that either no functional PEX2 protein is produced or that the truncated proteins are not correctly localized to the peroxisomal membrane. The mutation in patient 3 has been described before in homozygous (10) and compound heterozygous form (24, 25). Both homozygous patients described presented with the ZS phenotype. In one compound heterozygous patient the mutation was found in combination with a temperature-sensitive missense mutation (E55K), which led to a milder phenotype, whereas in another patient it was found in combination with an R125X mutation on the other allele. This patient was diagnosed with the severe ZS phenotype and died before 3 mo of age. In addition to this, one more patient was described lacking (one of) the transmembrane domains. This patient was homozygous for a del550C mutation, leading to a frameshift at amino acid 184 and termination seven amino acids downstream (26). The predicted protein sequence lacks both the second transmembrane domain and the zinc-binding domain and results in the ZS phenotype. These additional cases support our conclusions on patient 3 and 4.
Biochemical data are in agreement with the genotype in patients 3 and 4: DHAPAT activity, C26:0 β-oxidation and pristanic acid β-oxidation are clearly abnormal. Recent studies in fibroblasts of patients suffering from severe and mild forms of PBD have shown that DHAPAT activity, C26:0 β-oxidation and, to a lesser extent, pristanic acid β-oxidation correlate best with patients' survival (27).
The mutation found in patient 2 is predicted to result in a protein that lacks the zinc-binding domain but does contain the two transmembrane domains. This patient was described before as a patient with a “new type of peroxisomal disorder with variable expression in liver and fibroblasts”(16). When examined at 9 mo of age, no major abnormalities were found but after the 1st year of life neurodegenerative symptoms developed and at the age of 9 he was in a vegetative state. The patient died from pneumonia at the age of 13 y. Previous studies in fibroblasts of this patient did not reveal peroxisomal abnormalities with regard to de novo plasmalogen synthesis, DHAPAT activity and the presence of catalase in a particle bound form. However, these studies were done in the beginning of the 1990s, and since then more sensitive methods to assess peroxisomal functioning have been developed including immunofluorescence microscopy analysis using antibodies raised against catalase as well as the measurements of C26:0 and pristanic acid β-oxidation. When we reinvestigated the patient's fibroblasts using these methods we found clear abnormalities such as a reduced rate of pristanic acid β-oxidation and an abnormal catalase immunofluorescence pattern with both positive and negative cells (Table 2). This mosaic pattern is indicative of a peroxisomal biogenesis defect. Biochemical parameters such as VLCFA, phytanic acid and DHCA and THCA levels were also abnormal in plasma of the patient. In liver, catalase was localized in only 10% of peroxisomes (16). Although PEX2 RNA levels were decreased to 35%, in part of the fibroblasts peroxisomes were normally present. This suggests that the truncated form of PEX2 lacking the zinc-binding domain that is present may be localized correctly and is still (partly) active. This would imply that the zinc-binding domain is not obligatory for the activity of PEX2. A similar phenomenon was described before in another PEX2-deficient patient that had a deletion (642delG) leading to a frameshift at amino acid 214 leading to termination two amino acids downstream (26). This patient was diagnosed also with a mild phenotype (IRD). In this patient only fibroblasts were studied, which also showed a mosaic pattern of catalase staining (20% of the cells contained normal peroxisomes), and mildly reduced VLCFA β-oxidation and normal DHAPAT activity. It can be concluded from both our patient as well as the patient reported by Shimozawa et al. 2000 (26), that truncated PEX2 lacking the zinc-binding domain still displays some functional activity. The mutation found in patient 1, which changes the second cysteine residue (position 247) of the zinc-binding domain into arginine, however, seems to disagree with this postulate. This patient displays a severe phenotype and severely impaired biochemical parameters, indicative of a severe impairment in peroxisome biogenesis. A comparable mutation was found in a CHO-mutant (C246Y; the CHO PEX2 comprises 304 amino acids, one residue shorter than human PEX2) (28) which also did not show any catalase positive particles. Apparently, the complete absence of the zinc-binding domain is less deleterious for the functioning of PEX2, than a mutation within the domain itself. Because PEX2 RNA transcript levels in this patient were normal, this may be due to an increased instability of PEX2 protein due to this mutation. Unfortunately, no antibodies raised against full length PEX2 are available to study this possibility. Alternatively, it may be that the mutation in the zinc-binding domain, which is thought to function in the interaction with other proteins (for instance PEX10 (13)), causes an inhibiting effect on the function of these other proteins, thereby causing a severe impairment of peroxisome biogenesis. Such a “dominant negative” effect is not present in patients completely lacking the zinc-binding domain. CHO cell lines with missense mutations of two other cysteine residues in the zinc-binding domain have also been reported. A C258Y mutation has been described, which leads to a disturbed import of PTS1 proteins, whereas the PTS2 protein peroxisomal thiolase is normally imported (29). A CHO mutant with a C264S mutation was found to have a normal catalase import (30). Thus, not all cysteine mutations in this region lead to a severe defect.
Mutations in the other two zinc-binding domain-containing peroxins PEX10 and PEX12 have also been reported. In PEX10, one mutation leads to truncated PEX10 lacking the zinc-binding domain (31, 32). All patients homozygous for this mutation were diagnosed with the severe ZS phenotype. Moreover, all patients lacking the zinc-binding domain of PEX12 display a severe clinical phenotype and abnormal biochemical parameters (33), suggesting that in PEX12 the zinc-binding domain is indispensable for its function. Thus, regarding the zinc-binding domain, the genotype-phenotype correlation for PEX2 seems to be different from for other proteins, making the function of this domain in PEX2 unclear, and worthwhile to study in more detail.
Abbreviations
- PBD:
-
peroxisome biogenesis disorder
- ZS:
-
Zellweger syndrome
- IRD:
-
infantile Refsum disease
- DHAPAT:
-
dihydroxyacetonephosphate acyltransferase
- VLCFA:
-
very-long chain fatty acid
- DHCA:
-
dihydroxycholestanoic acid
- THCA:
-
trihydroxycholestanoic acid
- PTS:
-
peroxisomal targeting signal
- CG:
-
complementation group
References
Gould SJ, Raymond GV, Valle D 2001 The peroxisome biogenesis disorders. In: Scriver CR, Beaudet AL, Valle D, Sly WS (eds). The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill information Services Company, New York, 3181–3217.
Kelley RI, Datta NS, Dobyns WB, Hajra AK, Moser AB, Noetzel MJ, Zackai EH, Moser HW 1986 Neonatal adrenoleukodystrophy: new cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes. Am J Med Genet 23: 869–901
Poll-The BT, Saudubray JM, Ogier HA, Odievre M, Scotto JM, Monnens L, Govaerts LC, Roels F, Cornelis A, Schutgens RB 1987 Infantile Refsum disease: an inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy. Eur J Pediatr 146: 477–483
Barth PG, Gootjes J, Bode H, Vreken P, Majoie CB, Wanders RJ 2001 Late onset white matter disease in peroxisome biogenesis disorder. Neurology 57: 1949–1955
Heymans HS, Schutgens RB, Tan R, van den Bosch H, Borst P 1983 Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature 306: 69–70
Datta NS, Wilson GN, Hajra AK 1984 Deficiency of enzymes catalyzing the biosynthesis of glycerol-ether lipids in Zellweger syndrome. A new category of metabolic disease involving the absence of peroxisomes. N Engl J Med 311: 1080–1083
Gould SJ, Valle D 2000 Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet 16: 340–345
Subramani S 1996 Protein translocation into peroxisomes. J Biol Chem 271: 32483–32486
Terlecky SR, Legakis JE, Hueni SE, Subramani S 2001 Quantitative analysis of peroxisomal protein import in vitro. Exp Cell Res 263: 98–106
Shimozawa N, Tsukamoto T, Suzuki Y, Orii T, Shirayoshi Y, Mori T, Fujiki Y 1992 A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science 255: 1132–1134
Biermanns M, Gartner J 2000 Genomic organization and characterization of human PEX2 encoding a 35- kDa peroxisomal membrane protein. Biochem Biophys Res Commun 273: 985–990
Harano T, Shimizu N, Otera H, Fujiki Y 1999 Transmembrane topology of the peroxin, Pex2p, an essential component for the peroxisome assembly. J Biochem 125: 1168–1174
Okumoto K, Abe I, Fujiki Y 2000 Molecular anatomy of the peroxin Pex12p: ring finger domain is essential for Pex12p function and interacts with the peroxisome- targeting signal type 1-receptor Pex5p and a ring peroxin, Pex10p. J Biol Chem 275: 25700–25710
Reguenga C, Oliveira ME, Gouveia AM, Sa-Miranda C, Azevedo JE 2001 Characterization of the mammalian peroxisomal import machinery: Pex2p, Pex5p, Pex12p, and Pex14p are subunits of the same protein assembly. J Biol Chem 276: 29935–29942
Wanders RJ, Schutgens RB, Barth PG 1995 Peroxisomal disorders: a review. J Neuropathol Exp Neurol 54: 726–739
Mandel H, Espeel M, Roels F, Sofer N, Luder A, Iancu TC, Aizin A, Berant M, Wanders RJ, Schutgens RB 1994 A new type of peroxisomal disorder with variable expression in liver and fibroblasts. J Pediatr 125: 549–555
Ofman R, Wanders RJ 1994 Purification of peroxisomal acyl-CoA: dihydroxyacetonephosphate acyltransferase from human placenta. Biochim Biophys Acta 1206: 27–34
Wanders RJ, Denis S, Ruiter JP, Schutgens RB, van Roermund CW, Jacobs BS 1995 Measurement of peroxisomal fatty acid beta-oxidation in cultured human skin fibroblasts. J Inherit Metab Dis 18( suppl 1): 113–124
Heikoop JC, van Roermund CW, Just WW, Ofman R, Schutgens RB, Heymans HS, Wanders RJ, Tager JM 1990 Rhizomelic chondrodysplasia punctata. Deficiency of 3-oxoacyl-coenzyme A thiolase in peroxisomes and impaired processing of the enzyme. J Clin Invest 86: 126–130
Brul S, Westerveld A, Strijland A, Wanders RJ, Schram AW, Heymans HS, Schutgens RB, van den Bosch H, Tager JM 1988 Genetic heterogeneity in the cerebrohepatorenal (Zellweger) syndrome and other inherited disorders with a generalized impairment of peroxisomal functions. A study using complementation analysis. J Clin Invest 81: 1710–1715
Shimozawa N, Suzuki Y, Orii T, Moser A, Moser HW, Wanders RJ 1993 Standardization of complementation grouping of peroxisome-deficient disorders and the second Zellweger patient with peroxisomal assembly factor-1 (PAF-1) defect. Am J Hum Genet 52: 843–844
Jacobson A, Peltz SW 1996 Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem 65: 693–739
Maquat LE 1996 Defects in RNA splicing and the consequence of shortened translational reading frames. Am J Hum Genet 59: 279–286
Imamura A, Tsukamoto T, Shimozawa N, Suzuki Y, Zhang Z, Imanaka T, Fujiki Y, Orii T, Kondo N, Osumi T 1998 Temperature-sensitive phenotypes of peroxisome-assembly processes represent the milder forms of human peroxisome-biogenesis disorders. Am J Hum Genet 62: 1539–1543
Shimozawa N, Suzuki Y, Tomatsu S, Nakamura H, Kono T, Takada H, Tsukamoto T, Fujiki Y, Orii T, Kondo N 1998 A novel mutation, R125X in peroxisome assembly factor-1 responsible for Zellweger syndrome. Hum Mutat Suppl 1: S134–S136
Shimozawa N, Zhang Z, Imamura A, Suzuki Y, Fujiki Y, Tsukamoto T, Osumi T, Aubourg P, Wanders RJ, Kondo N 2000 Molecular mechanism of detectable catalase-containing particles, peroxisomes, in fibroblasts from a PEX2-defective patient. Biochem Biophys Res Commun 268: 31–35
Gootjes J, Mooijer PA, Dekker C, Barth PG, Poll-The BT, Waterham HR, Wanders RJ 2002 Biochemical markers predicting survival in peroxisome biogenesis disorders. Neurology 59: 1746–1749
Thieringer R, Raetz CR 1993 Peroxisome-deficient Chinese hamster ovary cells with point mutations in peroxisome assembly factor-1. J Biol Chem 268: 12631–12636
Huang Y, Ito R, Miura S, Hashimoto T, Ito M 2000 A missense mutation in the RING finger motif of PEX2 protein disturbs the import of peroxisome targeting signal 1 (PTS1)-containing protein but not the PTS2-containing protein. Biochem Biophys Res Commun 270: 717–721
Tsukamoto T, Shimozawa N, Fujiki Y 1994 Peroxisome assembly factor 1: nonsense mutation in a peroxisome- deficient Chinese hamster ovary cell mutant and deletion analysis. Mol Cell Biol 14: 5458–5465
Okumoto K, Itoh R, Shimozawa N, Suzuki Y, Tamura S, Kondo N, Fujiki Y 1998 Mutations in PEX10 is the cause of Zellweger peroxisome deficiency syndrome of complementation group B. Hum Mol Genet 7: 1399–1405
Warren DS, Wolfe BD, Gould SJ 2000 Phenotype-genotype relationships in PEX10-deficient peroxisome biogenesis disorder patients. Hum Mutat 15: 509–521
Chang CC, Gould SJ 1998 Phenotype-genotype relationships in complementation group 3 of the peroxisome-biogenesis disorders. Am J Hum Genet 63: 1294–1306
Acknowledgements
The authors thank Petra Mooijer and Conny Dekker for biochemical analyses in patient fibroblasts. Dr. Deprettere is gratefully acknowledged for his help in the diagnosis of one of the patients.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Prinses Beatrix Fonds, Grant 99.0220.
Rights and permissions
About this article
Cite this article
Gootjes, J., Elpeleg, O., Eyskens, F. et al. Novel Mutations in the PEX2 Gene of Four Unrelated Patients with a Peroxisome Biogenesis Disorder. Pediatr Res 55, 431–436 (2004). https://doi.org/10.1203/01.PDR.0000106862.83469.8D
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000106862.83469.8D
This article is cited by
-
Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene
Orphanet Journal of Rare Diseases (2011)
