
Abstract
Recent evidence strongly implicates the inflammatory response to intrauterine infection in the pathogenesis of neonatal brain and lung injury. We hypothesized that lung and brain injury in preterm infants occurs during a common developmental window of vulnerability as the result of an inflammatory response in different compartments. To determine whether inflammatory markers in these compartments are associated with bronchopulmonary dysplasia (BPD) or cranial ultrasound (CUS) abnormalities in infants <33 wk gestation age (GA) and <1501 g birth weight, we analyzed placental pathology and serum and cerebrospinal fluid (CSF) IL-6, IL-1β, and tumor necrosis factor-α (TNF-α) concentrations in 276 infants. Logistic regressions were performed stratified by GA. Histologic chorioamnionitis was significantly associated with BPD in infants ≤28 wk GA (OR 3.6, p = 0.027). Maternal stage of chorioamnionitis significantly correlated with severity of BPD. Presence of a fetal inflammatory response indicated by fetal vasculitis or elevated cytokines was not associated with the development of BPD. Serum IL-6 ≥17 pg/mL was associated with an abnormal CUS in infants >28 wk GA (OR 3.36, p = 0.023) but not ≤28 wk GA. CSF concentrations of IL-6 ≥6.5 pg/mL and TNF-α ≥3 pg/mL were associated with abnormal CUS in infants ≤28 wk GA (IL-6 OR 3.0; TNF-α OR 3.5; p < 0.05 each case) but not ≥28 wk GA. These data suggest that in infants ≤28 wks GA, BPD may be initiated by inflammatory mediators in amniotic fluid, but brain injury may involve variations in the systemic inflammatory response.
Similar content being viewed by others

Main
Preterm birth is the leading cause of perinatal morbidity and mortality in the world (1) and is the second leading cause of infant mortality in the United States. Preterm labor and preterm premature rupture of membranes account for 80% of preterm deliveries (2). There is accumulating evidence that intrauterine infection is a major factor in preterm labor and preterm premature rupture of the membranes, particularly in gestations of <30 wk duration (2). Intrauterine infection has also been implicated in the pathogenesis of adverse neonatal sequelae, including bronchopulmonary dysplasia (BPD), intraventricular hemorrhage (IVH), white matter damage (WMD) such as periventricular leukomalacia (PVL) and ventriculomegaly, and long-term disabilities such as cerebral palsy (3–5). It has been proposed that 23–32 wk gestation is a developmental period during which the lung and brain share a vulnerability to injury mediated by infection/inflammation (3, 4). During this period of saccular lung development (4) and predominance of the oxidative stress–sensitive preoligodendrocyte in the fetal brain (6–8), exposure of the fetus to intrauterine infection/ inflammation alters developmental signaling by mechanisms not completely understood.
Infection-induced up-regulation of inflammatory cytokines has been proposed to be the causative link between intrauterine infection and lung and brain injury (4, 5). Elevated concentrations of proinflammatory cytokines IL-6, IL-1β, IL-8, and tumor necrosis factor-α (TNF-α) in amniotic fluid (9), tracheal aspirates (10–15), blood (16–18), and brain tissue (19–21) have been associated with adverse outcomes in preterm infants. We propose that intrauterine infection–induced injury to the preterm lung and brain is initiated in utero, occurs during a developmental-dependent window of vulnerability, and is mediated by the inflammatory response in specific compartments. To our knowledge, no previous study has evaluated the inflammatory response in all three relevant compartments (fetoplacental unit, fetal circulation, and CNS) simultaneously. The purpose of this prospective study was to determine whether evidence of inflammation in placenta, blood, or cerebrospinal fluid (CSF) increases the risk for BPD defined as oxygen dependence at 28 d or cranial ultrasound (CUS) abnormalities (IVH and/or WMD) in very low birth weight infants. To define further the developmental period of vulnerability, we compared the association of inflammatory markers to outcomes in infants ≤28 wk gestation and in more mature infants.
METHODS
Sample.
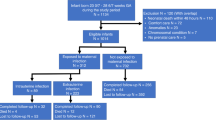
Inborn infants who had GA <33 wk and birth weight <1501 g and were admitted to the neonatal intensive care units at the University of Maryland Medical System and Mercy Medical Center (Baltimore, MD, U.S.A.) between June 1999 and June 2002 were eligible for this study. Infants with congenital brain/neural tube defects or confirmed congenital TORCH infections were excluded. Parental consent was obtained, and the University of Maryland, Baltimore, and Mercy Medical Center Institutional Review Boards approved the study protocol.
Because CSF samples were the most limiting in this study, we performed a sample size analysis for CUS abnormalities with and without elevated CSF IL-6. We determined the sample size that would provide 80% power to reject the null hypothesis of equal CUS abnormality rates in infants with and without elevated CSF IL-6 when the rates are in fact 65% with and 40% without elevated CSF IL-6. For achieving 80% power with a two-sided α level of 0.05 and a 1:1 ratio of infants in the two groups, 85 infants were required in each group. Sufficient subjects who had a lumbar puncture performed as part of a sepsis evaluation were enrolled to achieve the determined sample size.
Serum and CSF cytokines.
After informed consent was obtained, cord blood (2 mL) or venous blood (1 mL) obtained within 12 h of delivery were drawn aseptically, and the serum was separated, aliquoted, and stored at −80°C for cytokine measurements. Because cytokine levels rapidly decline after birth (22, 23) and for specifically evaluating cytokine concentrations in response to fetal events rather than postnatal events, blood sampling was limited to <12 h of age. Two thirds of the serum samples were cord and the remainder were venous samples.
For enrolled infants for whom a lumbar puncture was performed as standard neonatal intensive care unit practice as part of a sepsis evaluation within 72 h after delivery, sufficient CSF was removed for routine clinical studies (bacterial culture, glucose and protein, and cell count and differential) and an additional 1.0 mL was removed for cytokine measurements. Lumbar punctures were not performed for study purposes only. CSF was obtained from 173 (63%) study subjects at a median age of 14.5 h (range, 1–71 h). Serum and CSF samples were centrifuged at 1000 ×g to pellet cells, and the supernatants were stored at −80°C until analysis. Cytokines (TNF-α, IL-1β, and IL-6) were measured in duplicate in serum and CSF samples using standard two-antibody ELISA with commercial antibody pairs and recombinant standards (Endogen, Boston, MA, U.S.A.) as previously described (24). A curve was fit to the standards using a computer program (Softpro: Molecular Devices), and cytokine concentrations from each sample were calculated from the standard curve. Assay sensitivities were 3, 0.78, and 1.5 pg/mL for TNF-α, IL-1β, and IL-6, respectively.
Placental pathology.
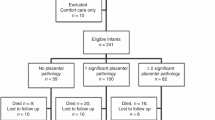
Placental studies were performed on 230 (83%) of the 276 study subjects. There were no differences in incidence of BPD and CUS abnormalities in infants with and without placental examination. Formalin-fixed, paraffin-embedded, and hematoxylin- and eosin-stained sections of umbilical cord, membrane roll, placental disc near the cord insertion site as well as the midpoint between cord insertion, and periphery of the placental disc were prepared (25). The sections were reviewed in a blinded manner according to a standard protocol (26). Histologic chorioamnionitis was separated into maternal and fetal involvement and assigned a stage according to the scheme proposed by Redline et al. (26). Maternal stages were as follows: stage 1, neutrophils in sub-chorionic plate; stage 2, neutrophils in the chorionic laevae; stage 3, neutrophils in the chorion laevae and adjacent amnion; 4, neutrophil karyorrhexis or eosinophilia of basement membrane without amnionic epithelial sloughing; and stage 5, neutrophils plus amnionic epithelial sloughing. Fetal vasculitis was defined as polymorphonuclear infiltration of the chorionic vessels or umbilical cord (26). Fetal stages were as follows: stage 1, neutrophils in chorion vessels only; stage 2, neutrophils in umbilical vein wall; stage 3, neutrophils in all three umbilical vessels; stage 4, neutrophils in Wharton's jelly; and stage 5, concentric umbilical perivasculitis.
CUS scans.
Serial CUS examinations were performed on 273 (99%) subjects using a high-frequency variable bandwidth transducer (5.5– 8.5 MHz; Acuson Sequoia 512, Mountain View, CA, U.S.A.) for detection of WMD and IVH at the following time points:1) between 3 and 7 postnatal days, 2) at 28–30 postnatal days, and 3) at 34–36 wk postmenstrual age (PMA) or pretransfer or predischarge and as clinically indicated. Because of practical constraints, obtaining scans before performing the lumbar puncture was not attempted. CUSs were reviewed and classified as normal or abnormal with the following diagnoses:1) germinal matrix/IVH (27), 2) transient periventricular echodensities (resolution without progressing to echolucencies) (28), 3) persistent periventricular echodensities (echodensities present on two or more scans without resolution or progression to echolucencies) (29), 4) periventricular echolucencies, and 5) ventricular enlargement. WMD included periventricular echodensities and echolucencies and ventricular enlargement. A single radiologist at each site read scans. For determining interrater agreement, both radiologists read a subset of scans. The agreement between radiologists was 75% with a κ of 0.53 for presence of any CUS abnormality, 62.5% with a κ of 0.45 for presence and severity of IVH, and 87.5% with a κ of 0.6 for WMD lesions. For scans with different interpretations, the primary reader's interpretation was used.
BPD
A recent National Institutes of Health/National Institute of Child Health and Human Development/ORD (Office of Rare Diseases) workshop on BPD proposed the following defini-tions for BPD:1) no BPD, supplemental O2 <28 d; 2) mild BPD, supplemental O2 for 28 d but breathing room air at 36 wk PMA; 3) moderate BPD, need for <30% fraction of inspired oxygen at 36 wk PMA; and 4) severe BPD, need for ≥30% fraction of inspired oxygen or positive pressure support at 36 wk PMA (30). For pulmonary outcomes in this study, the moderate and severe stages of BPD were combined.
CLINICAL VARIABLES
The following variables were evaluated:1) demographic variables; 2) obstetric variables including maternal drug use, parity, multiple gestations, preterm labor, preterm premature rupture of membranes, pregnancy-induced hypertension (defined as a systolic blood pressure >140 mm Hg and a diastolic blood pressure >90 mm Hg plus proteinuria with or without edema), clinical chorioamnionitis (maternal temperature ≥38°C and two of the following: uterine tenderness, malodorous vaginal discharge, fetal tachycardia >160 bpm, or maternal white blood cell count >15,000), antepartum hemorrhage, use of medications (MgSO4, other tocolytics, antenatal steroids, antibiotics), evidence of fetal heart rate abnormalities, and mode of delivery; and 3) neonatal factors, including Apgar scores at 1 and 5 min, need for resuscitation, birth weight, gestational age by best obstetric estimate (31), length, head circumference, size for gestational age, sex, administration of exogenous surfactant, patent ductus arteriosus, early or late culture-confirmed sepsis, necrotizing enterocolitis, hypotension (defined as mean blood pressure <10th percentile for weight and postnatal age sustained for ≥2 consecutive h during the first 96 h of age (29), duration of mechanical ventilation and supplemental oxygen, and hypothyroxinemia (total T4 <5.3 μg/dL) (32). Admission complete blood count and differential, C-reactive protein on admission and 12–24 h age, blood and CSF bacterial culture results, CSF cell count and differential, and CSF glucose and protein concentrations were recorded.
STATISTICAL ANALYSIS
The t test was used to compare continuous variables, and the χ2 or Fisher exact test was used to compare categorical variables. Univariate odds ratios (ORs) and 95% confidence intervals (CIs) were calculated for all variables for outcomes BPD and CUS abnormalities. Because the range of cytokine concentrations was large, for analyses, cytokine data expressed as pg/mL were dichotomized using the median of serum and CSF IL-6 concentrations and serum IL-1β concentrations for the entire sample and the lower cutoff of ELISA sensitivity for TNF-α concentrations and CSF IL-1β concentrations. Stepwise logistic regressions were performed to identify the clinical variables significantly associated with each inflammatory variable (histologic chorioamnionitis, fetal vasculitis, white blood cell count, absolute neutrophil count, and serum and CSF cytokine concentrations). The contribution of each inflammatory variable to risk for BPD or CUS abnormalities was then evaluated in logistic regression models that included the potential confounders identified in the stepwise analyses. All multivariate analyses were performed with and without stratification by GA ≤28 wk and >28 wk. For determining whether there were interactions of postnatal variables with the inflammatory variables for each outcome, interaction terms were generated and included in regression models. All statistical analyses were performed using Stata (Stata Corp., College Station, TX, U.S.A.). A p < 0.05 was considered significant.
RESULTS
Cohort characteristics and outcomes.
A total of 276 infants were enrolled. The clinical characteristics and primary outcomes are presented in Table 1. The mean (SD) GA of the cohort was 27.6 ± 2.5 wk and mean birth weight was 987 ± 281 g. Overall mortality was 6.5% with 14 deaths occurring before the subjects reached 36 wk PMA. Of the survivors, 37% had mild BPD and 21% had moderate to severe BPD as defined by the recent National Institutes of Health criteria (30). The majority of infants who developed BPD were ≤28 wk GA. CUS abnormalities were documented on one or more serial scans in 138 (50%) of the entire cohort. The CUS diagnoses for the entire cohort included IVH grade 1, 20%; grade 2, 10%; grade 3, 8.7%; grade 4, 5.4%; PVL, 7.6%; and ventriculomegaly, 13%. The overall frequency of CUS abnormalities was higher in infants ≤ 28 wk (99 of 175; 57%) than in infants >28 wk (39 of 101; 39%; p = 0.006). The incidence of severe IVH (≥grade 3) was 22-fold higher in infants ≤28 wk (38 of 175; 22%) than in infants >28 wk (1 of 101; 1%; p < 0.001;Table 1). There were no differences in age of lumbar puncture between infants with and without CUS abnormalities. CSF pleocytosis (>40 white blood cells corrected for red blood cells) was detected in 15 (9%) CSF samples. Only one CSF was culture positive for Staphylococcus epidermidis.
Characteristics of infants with and without chorioamnionitis.
Histologic chorioamnionitis was present in 147 (64%) of 230 placentas submitted for pathologic examination. The incidence of histologic chorioamnionitis was inversely related to GA (≤28 wk, 72.5%; >28 wk 48%; p = 0.001; Table 2). All but one case of fetal vasculitis occurred in association with histologic chorioamnionitis. Clinical variables associated with an increased risk for histologic chorioamnionitis are presented in Table 3. The clinical diagnosis of chorioamnionitis was made in only 40% of cases of histologic chorioamnionitis, indicating that the majority of intrauterine infections are subclinical. Infants who were exposed to histologic chorioamnionitis were more likely to receive >7 d of mechanical ventilation and ≥28 d of supplemental oxygen.
Because 99% of placentas with fetal vasculitis had membrane inflammation, the specific contributions of maternal and fetal involvement to risk for a systemic inflammatory response were assessed in logistic regression analyses in which the sample of infants was restricted to those with chorioamnionitis without fetal involvement (n =34) to assess chorioamnionitis alone, or to those with presence of fetal vasculitis. As shown in Table 4, inflammation of the fetal membranes and cord was significantly associated with blood and CSF inflammatory variables. The admission absolute neutrophil count was higher in infants who were exposed to histologic chorioamnionitis and/or fetal vasculitis. Elevated serum IL-6 concentration ≥17 pg/mL and IL-1β concentration ≥5.4 were associated with increased risk for chorioamnionitis and fetal vasculitis. In contrast, serum TNF-α concentrations >3 pg/mL were significantly associated with umbilical cord but not fetal membrane inflammation. Of the three cytokines measured in CSF, only elevated IL-1β was associated with fetal vasculitis, and none was associated with histologic chorioamnionitis. Serum IL-6 concentrations were significantly correlated with paired serum IL-1β (r2 = 0.803) and TNF-α (r2 = 0.481) concentrations (p < 0.05 each case). CSF IL-6 concentration correlated with CSF IL-1β concentration (r2 = 0.402) but not with CSF TNF-α concentrations. Serum cytokine concentrations did not correlate with CSF cytokine concentrations even when the analyses were restricted to paired samples obtained at <24 h of age.
Inflammatory indicators and risk for BPD.
Variables associated with BPD in univariate analyses are shown in Table 5. In a logistic regression model adjusted for confounding variables including GA, histologic chorioamnionitis was associated with BPD (OR 3.13, 1.2– 8.2; p = 0.02). After stratification by gestational age, histologic chorioamnionitis remained significantly associated with BPD in infants ≤ 28 wk GA, but not in infants >28 wk GA (Table 6). Maternal stage of chorioamnionitis was significantly correlated with severity of BPD as defined by duration of supplemental oxygen and ventilation (Fig. 1). Forty percent of infants who developed moderate-severe BPD were exposed to long standing or necrotizing amnionitis (maternal stages 4 and 5) compared with 34% of infants who developed mild BPD and 19% of infants without BPD. The presence of a fetal inflammatory response indicated by fetal vasculitis or elevated inflammatory cytokines in serum and CSF was not associated with the development of BPD. There were no significant interactions of any inflammatory variable with postnatal clinical variables such as duration of mechanical ventilation or sepsis for BPD risk.

Relationship of BPD severity and maternal stage of chorioamnionitis. Percentage of study infants with and without BPD (30) with no histologic chorioamnionitis (□) or maternal involvement by stages (see “Methods”).
Inflammatory indicators and risk for CUS abnormalities.
Clinical variables associated with an increased risk for CUS abnormalities are shown in Table 7. After adjustment for confounding variables with or without stratification for GA, neither histologic chorioamnionitis nor fetal vasculitis was associated with CUS abnormalities (Table 8). In regression models including GA as a variable, serum IL-6 ≥ 17 pg/mL and CSF IL-6 ≥ 6.5 pg/mL were associated with CUS abnormalities (serum IL-6 OR 1.88, 95% CI 1.01–3.5; p = 0.045; CSF IL-6 OR 2.13, 95% CI 1.14– 4.0; p = 0.018). After stratification by GA and adjustment for confounding, elevated serum IL-6 was associated with an abnormal CUS in infants >28 wk GA but not in infants ≤28 wk GA (Table 8). Because elevated cord serum IL-6 concentration is reported to be associated with decreased blood pressure in preterm infants (33), we assessed whether systemic hypotension was associated with serum IL-6 in the current cohort. Elevated serum IL-6 was not associated with hypotension in either GA subgroup. Moreover, there was no significant interaction of serum IL-6 and hypotension in either subgroup. When stratified by GA, CSF concentrations of IL-6 ≥6.5 pg/mL and TNF-α ≥3 pg/mL were associated with abnormal CUS in infants ≤28 wk GA but not in infants ≥28 wk GA. Although elevated serum and CSF IL-1β concentrations were associated with fetal vasculitis, they were not significantly associated with CUS abnormalities. An elevated CSF cell count was not significantly associated with placental inflammation or abnormal CUS. There were no significant interactions of any inflammatory variable with postnatal clinical variables for CUS risk.
DISCUSSION
In this study, we demonstrated that placental inflammation is associated with BPD and that elevated CSF IL-6 and TNF-α are associated with CUS abnormalities in infants ≤28 wk GA. There are a number of possible study limitations, however. First, because lumbar punctures were performed only in infants with suspected sepsis, there may have been a selection bias toward infants with greater exposure to inflammation and possibly higher CSF cytokines than would be present in the whole cohort. Second, although serial scans were performed to maximize detection of lesions, some lesions may have been missed. Third, the concordance of radiologists was moderate but within the reported concordance for neonatal CUS interpretations (34). Fourth, the timing of the first scan at 3–7 d of age did not allow us to determine the timing of IVH. Finally, because the sample size of infants >28 wk with BPD was likely too small to draw conclusions about the relationship of chorioamnionitis and BPD, our discussion is limited to the findings in infants ≤28 wk GA.
Determination of the risk factors for BPD and development of effective intervention strategies have been hampered by differences in diagnostic criteria among studies such as time of assessment (28 d versus 36 wk PMA) (35, 36), inclusion of radiographic criteria (35, 37), and tracheal aspirate cytology (38). Using diagnostic criteria for BPD that were recommended by a recent National Institute of Child Health and Human Development/National Heart, Lung, and Blood Institute/ORD workshop (30), the present study demonstrated that infants who were ≤28 wk GA and were exposed to histologic chorioamnionitis had an increased risk for BPD (oxygen dependence at 28 d). Our observations agree with the findings of Watterberg et al. (10) that exposure to histologic chorioamnionitis is associated with increased risk for BPD. In contrast, in a case-control study, Van Marter et al. (39) observed that histologic chorioamnionitis alone was associated with decreased risk for chronic lung disease (oxygen dependence at 36 wk PMA) but was associated with an increased risk for chronic lung disease in infants who were exposed to prolonged mechanical ventilation or postnatal sepsis (combined clinical and culture-proven sepsis). In a retrospective study, Redline et al. (40) observed that histologic chorioamnionitis tended to be increased in white infants and decreased in black infants with oxygen dependence at 36 wk PMA. In our prospective study, logistic regressions including interaction terms for clinical variables did not demonstrate significant interactions of histologic chorioamnionitis with any clinical variable for BPD risk.
The frequency of amnionitis (maternal stages 3, 4, and 5) was higher in infants with more severe BPD. The secretion of inflammatory cytokines into amniotic fluid in response to intrauterine infection by amniotic epithelium (41, 42) or by infiltrating phagocytic cells may initiate lung injury in utero. Amniotic fluid concentrations of IL-1β, IL-6, TNF-α, and IL-8 are higher in pregnancies that produce infants who develop BPD than in pregnancies that produce infants without BPD (9). Increased concentrations of IL-6, IL-1β, and IL-8 are detected in tracheal aspirates of preterm infants on the first day of life associated with prolonged rupture of the membranes (43) and histologic chorioamnionitis (10, 11, 44), respectively. In transgenic mice, overexpression of TNF-α, TGF-β, IL-6, or IL-11 inhibits alveolarization, suggesting that prolonged exposure of the preterm lung to a proinflammatory environment may contribute to abnormal septation (45). Taken together, these observations suggest that intrauterine infection–mediated fetal lung injury in infants ≤28 wk is the result of direct effects of proinflammatory cytokines on the developing distal airways. Airway injury/inflammation may be augmented in infants ≤ 28 wk by postnatal exposure to mechanical ventilation, oxygen, and infection. More mature infants with intrauterine inflammation are less likely to be exposed to these risk factors.
The presence of a fetal inflammatory response characterized by fetal vasculitis or elevated serum IL-6 was not significantly associated with BPD. The lack of an association of the fetal inflammatory response with risk for BPD in the present study differs from previous reports (16, 17, 46). Because our study and others (44) have demonstrated that indicators of the fetal inflammatory response (fetal vasculitis, elevated serum IL-6) are highly correlated with histologic chorioamnionitis (inflammation of fetal membranes), it is difficult to distinguish the independent contribution of the different compartments to outcome risk. The strength of the current study is the simultaneous assessment of inflammation in all relevant compartments.
Previous epidemiologic and placental pathology studies have identified intrauterine infection as a significant independent predictor of neonatal CUS abnormalities and later neurologic impairment including cerebral palsy. Preterm infants who are exposed to chorioamnionitis have a 2- to 4-fold increased risk of IVH (18, 47–50) and a 6-fold increased risk for PVL (51, 52). WMD occurs in 65% of preterm infants who are born to mothers with culture-positive amniotic fluid or histologic chorioamnionitis compared with 9% of infants who are born to mothers without evidence of intrauterine infection (9). Fetal vasculitis significantly increases the risk for early (≤10 d) and late (>10 d) PVL in a large multicenter cohort of very low birth weight infants (34). The presence of nonocclusive chorionic plate thrombi in placentas with chorioamnionitis is associated with neurologic impairment in very low birth weight infants at 20 mo of age (26), suggesting that placental inflammation activated coagulation may contribute to brain injury in utero.
In support of the “cytokine hypothesis of brain injury” (5), we provide evidence that the increased risk for CUS abnormalities in infants ≤28 wk GA is associated with an increased expression of IL-6 and TNF-α in the CNS compartment. Because CUS scans were not performed before the CSF was obtained, we cannot determine whether the proinflammatory cytokines were released in response to injury or directly contributed to brain injury. Because of the lack of correlation of paired serum and CSF cytokines, it is unlikely that serum-derived cytokines diffusing across the immature blood-brain barrier are a major source of the elevated CSF cytokines.
The preterm brain may be particularly vulnerable to the inflammatory/cytotoxic effects of cytokines. The process of myelination may be particularly susceptible to disruption by cytokine exposure. IL-6 shifts the development of the oligodendrocyte precursor cell in vitro from becoming myelin-producing oligodendrocytes to astrocytes (53). This may explain, in part, the astrocytosis in WMD (5). Before 28 wk GA, the white matter is mostly populated with the oxidative injury–sensitive preoligodendrocyte (7), suggesting that this period is particularly sensitive to inflammation-mediated injury. Vascular developmental immaturity may increase the susceptibility of the preterm brain to ischemia as a result of cytokine-activated coagulation/thrombosis pathways (26, 48), suggesting a convergence of the “vascular hypothesis” (8) and the “cytokine hypothesis” (5). Rupture of the ependyma by IVH may expose the adjacent white matter to CSF cytokines (48).
In the current study, elevated serum IL-6 was associated with an increased risk for CUS abnormalities in infants >28 wk GA. Although cord serum IL-6 concentration is associated with decreased blood pressure in preterm infants (33), suggesting that the risk for brain injury in these more mature infants exposed to IL-6 may be due, in part, to systemic and cerebrovascular hemodynamic disturbances, we did not observe an association of serum IL-6 with systemic hypotension. Alternatively, because there are bidirectional interactions of systemic and CNS cytokines, the brain may be injured by cytokines generated outside the CNS. Systemic sepsis or subcutaneously administered endotoxin induces brain IL-1 and IL-6 in mice (54). In juvenile rhesus monkeys, i.v. administration of IL-1β induces brain-derived IL-6 in CSF, suggesting that the brain responds to peripheral activation. Furthermore, systemic inflammatory cytokines may exacerbate injury mediated by excitatory neurotransmitters such as glutamate released in response to hypoxiaischemia (55, 56). In a model of ibotenate-induced excitotoxic brain injury in newborn mice, systemic pretreatment with IL-1β, IL-6, or TNF-α exacerbates ibotenate-induced brain lesions (56). Further investigation will be needed to determine the relative contribution of inflammation in the circulatory and CNS compartments to brain injury during different stages of development.
Nelson et al. (57) found no correlation of inflammatory cytokines measured at 2–3 postnatal d with later diagnosis of spastic cerebral palsy in a cohort of preterm infants. However, it remains to be determined whether cytokine measurements in blood or CSF obtained soon after birth and therefore largely reflecting intrauterine conditions will be markers for later neurosensory impairment. Because intrauterine infection/ inflammation is a major factor in preterm birth and adverse sequelae in gestations of ≤28 wk, intervention strategies should focus on early identification of high-risk women and the development of drugs to modulate the inflammatory response (55).
Abbreviations
- BPD:
-
bronchopulmonary dysplasia
- CI:
-
confidence interval
- CSF:
-
cerebrospinal fluid
- CUS:
-
cranial ultrasound
- GA:
-
gestational age
- IVH:
-
intraventricular hemorrhage
- OR:
-
odds ratio
- PMA:
-
postmenstrual age
- PVL:
-
periventricular leukomalacia
- TNF-α:
-
tumor necrosis factor-α
- WMD:
-
white matter damage
References
Gomez R, Ghezzi F, Romero R, Munoz H, Tolosa JE, Rojas I 1995 Premature labor and intra-amniotic infection. Clin Perinatol 22: 281–342.
Goldenberg RL, Hauth JC, Andrews WW 2000 Intrauterine infection and preterm delivery. N Engl J Med 342: 1500–1507.
Vigneswaran R 2000 Infection and preterm birth: evidence of a common causal relationship with bronchopulmonary dysplasia and cerebral palsy. J Paediatr Child Health 36: 293–296.
Jobe AH, Ikegami M 2001 Antenatal infection/inflammation and postnatal lung maturation and injury. Respir Res 2: 27–32.
Dammann O, Leviton A 1998 Infection remote from the brain, neonatal white matter damage, and cerebral palsy in the preterm infant. Semin Pediatr Neurol 5: 190–201.
Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ 1998 Maturation-dependent vulnerability of oligodentrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18: 6241–6253.
Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC 2001 Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21: 1302–1312.
Volpe JJ 2001 Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50: 553–562.
Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi J-H, Kim I-O 1997 Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1β, and tumor necrosis factor-α), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol 177: 19–26.
Watterberg KL, Demers LM, Scott SM, Murphy S 1996 Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 97: 210–215.
Kwong KY, Jones CA, Cayabyab R, Lecart C, Stotts CL, Randhawa I, Ramanathan R, Khuu N, Minoo P, deLemos RA 1998 Differential regulation of IL-8 by IL-1β and TNFα in hyaline membrane disease. J Clin Immunol 18: 71–80
Kotecha S, Wilson L, Wangoo A, Silverman M, Shaw RJ 1996 Increase in interleukin (IL)-1β and IL-6 in bronchoalveolar lavage fluid obtained from infants with chronic lung disease of prematurity. Pediatr Res 40: 250–256.
Rindfleisch MS, Hasday JD, Taciak V, Broderick K, Viscardi RM 1996 Potential role of interleukin-1 in the development of bronchopulmonary dysplasia. J Interferon Cytokine Res 16: 365–373.
Groneck P, Goetze-Speer B, Speer CP 1996 Inflammatory bronchopulmonary response of preterm infants with microbial colonisation of the airways at birth. Arch Dis Child Fetal Neonatal Ed 74:F51–F55.
Patterson AM, Taciak V, Lovchik J, Fox RE, Campbell AB, Viscardi RM 1998 Ureaplasma urealyticum respiratory tract colonization is associated with an increase in IL-1β and TNF-α relative to IL-6 in tracheal aspirates of preterm infants. Pediatr Infect Dis J 17: 321–328.
Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM 1998 The fetal inflammatory response syndrome. Am J Obstet Gynecol 179: 194–202.
Yoon BH, Romero R, Kim KS, Park JS, Ki SH, Kim BI, Jun JK 1999 A systemic fetal inflammatory response and the development of bronchopulmonary dysplasia. Am J Obstet Gynecol 181: 773–779.
Tauscher MK, Berg D, Brockmann M, Seidenspinner S, Speer CP, Groneck P 2003 Association of histologic chorioamnionitis, increased levels of cord blood cytokines, and intracerebral hemorrhage in preterm neonates. Biol Neonate 83: 166–170.
Yoon BH, Romero R, Kim CJ, Koo JN, Choe G, Syn HC, Chi JGC 1997 High expression of tumor necrosis factor-α and interleukin-6 in periventricular leukomalacia. Am J Obstet Gynecol 177: 406–411.
Deguchi K, Mizuguchi M, Takashima S 1996 Immunohistochemical expression of tumor necrosis factor alpha in neonatal leukomalacia. Pediatr Neurol 14: 13–16.
Deguchi K, Oguchi K, Takashima S 1997 Characteristic neuropathology of leukomalacia in extremely low birth weight infants. Pediatr Neurol 16: 296–300.
Messer J, Eyer D, Donato L, Gallati H, Matis J, Simeoni U 1996 Evaluation of interleukin-6 and soluble receptors of tumor necrosis factor for early diagnosis of neonatal infection. J Pediatr 129: 574–580.
Dammann O, Phillips TM, Allred EN, O'Shea TM, Paneth N, Van Marter LJ, Bose C, Ehrenkranz RA, Bednarek FJ, Naples M, Leviton A 2001 Mediators of fetal inflammation in extremely low gestational age newborns. Cytokine 13: 234–239.
Jiang Q, Cross AS, Singh IS, Chen TT, Viscardi RM, Hasday JD 2000 Febrile core temperature is essential for optimal host defense in bacterial peritonitis. Infect Immun 68: 1265–1270.
Langston C, Kaplan C, Macpherson T, Manci E, Peevy K, Clark B, Murtagh C, Cox S, Glenn G 1997 Practice guideline for examination of the placenta. Arch Pathol Lab Med 121: 449–476.
Redline RW, Wilson-Costello D, Borawski E, Fanaroff AA, Hack M 1998 Placental lesions associated with neurologic impairment and cerebral palsy in very low-birth-weight infants. Arch Pathol Lab Med 122: 1091–1098.
Papile L-A, Burstein J, Burstein R, Koffler H 1978 Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. J Pediatr 92: 529–534.
Dammann O, Leviton A 1997 Duration of transient hyperechoic images of white matter in very-low-birthweight infants: a proposed classification. Dev Med Child Neurol 39: 2–5.
Leviton A, Paneth N 1990 White matter damage in preterm newborns—an epidemiologic perspective. Early Hum Dev 24: 1–22.
Jobe AH, Bancalari E 2001 Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163: 1723–1729.
Donovan EF, Tyson JE, Ehrenkranz RA, Verter J, Wright LL, Korones SB, Bauer CR, Shankaran S, Stoll BJ, Fanaroff AA, Oh W, Lemons JA, Stevenson DK, Papile LA 1999 Inaccuracy of Ballard scores before 28 weeks' gestation. National Institute of Child Health and Human Development Neonatal Research Network. J Pediatr 135: 147–152.
Leviton A, Paneth N, Reuss ML, Susser M, Allred EN, Dammann O, Kuban K, Van Marter LJ, Pagano M 1999 Hypothyroxinemia of prematurity and the risk of cerebral white matter damage. J Pediatr 134: 706–711.
Yanowitz TD, Jordan JA, Gilmour CH, Towbin R, Bowen A, Roberts JM, Brozanski BS 2002 Hemodynamic disturbances in premature infants born after chorioamnionitis: association with cord blood cytokine concentrations. Pediatr Res 51: 310–316.
Leviton A, Paneth N, Reuss ML, Susser M, Allred EN, Dammann O, Kuban K, Van Marter LJ, Pagano M 1999 Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Pediatr Res 46: 566–575.
Bancalari E, Abdenour GE, Feller R, Gannon J 1979 Bronchopulmonary dysplasia: clinical presentation. J Pediatr 95: 819–829.
Shennan AT, Dunn MS, Ohlsson A, Lennox K, Hoskins EM 1988 Abnormal pulmonary outcomes in premature infants: prediction from oxygen requirement in the neonatal period. Pediatrics 82: 527–532.
Toce SS, Farrell PM, Leavitt LA, Samuels DP, Edwards DK 1984 Clinical and roentgenographic scoring systems for assessing bronchopulmonary dysplasia. Am J Dis Child 138: 581–585.
Viscardi RM, Broderick K, Sun CCJ, Yale-Loehr A, Hessamfar A, Taciak V, Burke KC, Koenig KB, Idell S 1992 Disordered pathways of fibrin turnover in lung lavage of premature infants with respiratory distress syndrome. Am Rev Respir Dis 146: 492–499.
van Marter LJ, Dammann O, Allred EN, Leviton A, Pagano M, Moore M, Martin C, Developmental Epidemiology Network Investigators 2002 Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr 140: 171–176.
Redline RW, Wilson-Costello D, Hack M 2002 Placental and other perinatal risk factors for chronic lung disease in very low birth weight infants. Pediatr Res 52: 713–719.
Fortunato SJ, Menon R, Swan KF, Menon R 1996 Inflammatory cytokine (interleukins 1, 6, and 8 and tumor necrosis factor-α) release from cultured human fetal membranes in response to endotoxic lipopolysaccharide mirrors amniotic fluid concentrations. Am J Obstet Gynecol 174: 1855–1862.
Reisenberger K, Egarter C, Knofler M, Schiebel I, Gregor H, Hirschl AM, Heinze G, Husslein P 1998 Cytokine and prostaglandin production by amnion cells in response to the addition of different bacteria. Am J Obstet Gynecol 178: 50–53.
Grigg JM, Barber A, Silverman M 1992 Increased levels of bronchoalveolar lavage fluid interleukin-6 in preterm ventilated infants after prolonged rupture of membranes. Am Rev Respir Dis 145: 782–786.
De Dooy J, Colpaert C, Schuerwegh A, Bridts C, Van Der Planken M, Ieven M, De Clerck L, Stevens W, Mahieu L 2003 Relationship between histologic chorioamnionitis and early inflammatory variables in blood, tracheal aspirates, and endotracheal colonization in preterm infants. Pediatr Res 54: 113–119.
Jobe AH 1999 The new BPD: an arrest of lung development. Pediatr Res 46: 641–643.
Matsuda T, Nakajima T, Hattori S, Hanatani K, Fukazawa Y, Kobayashi K, Fujimoto S 1997 Necrotizing funisitis: clinical significance and association with chronic lung disease in premature infants. Am J Obstet Gynecol 177: 1402–1407.
Yoon BH, Romero R, Kim CJ, Jun JK, Gomez R, Choi JH, Syn HC 1995 Amniotic fluid interleukin-6: a sensitive test for antenatal diagnosis of acute inflammatory lesions of preterm placenta and prediction of perinatal morbidity. Am J Obstet Gynecol 172: 960–970.
Dammann O, Leviton A 1997 Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42: 1–8.
Hansen A, Developmental Epidemiology Network Investigators 1998 The correlation between placental pathology and intraventricular hemorrhage in the preterm infant. Pediatr Res 43: 15–19.
De Felice C, Toti P, Laurini RN, Stumpo M, Picciolini E, Todros T, Tanganelli P, Buoncocore G, Bracci R 2001 Early neonatal brain injury in histologic chorioamnionitis. J Pediatr 138: 101–104.
Perlman JM, Risser R, Broyles RS 1996 Bilateral cystic periventricular leukomalacia in the premature infant: associated risk factors. Pediatrics 97: 822–827.
Resch B, Vollaard E, Maurer U, Haas J, Rosegger H, Muller W 2000 Risk factors and determinants of neurodevelopmental outcome in cystic periventricular leukomalacia. Eur J Pediatr 159: 663–670.
Kahn MA, de Villis J 1994 Regulation of an oligodendrocyte progenitor cell line by the interleukin-6 family of cytokines. Glia 12: 87–98.
Meyer TA, Wang JJ, Tiao GM, Ogle CK, Fischer JE, Hasselgren P-O 1997 Sepsis and endotoxaemia in mice stimulate the expression of interleukin-1 and interleukin-6 in the central nervous system. Clin Sci (Lond) 92: 519–525.
Dammann O, Leviton A 2000 Brain damage in preterm newborns: biological response modification as a strategy to reduce disabilities. J Pediatr 136: 433–438.
Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P 2000 Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol 47: 54–63.
Nelson KB, Grether JK, Dambrosia JM, Walsh E, Kohler S, Satyanarayana G, Nelson PG, Dickens BF, Phillips TM 2003 Neonatal cytokines and cerebral palsy in very preterm infants. Pediatr Res 53: 600–607.
Author information
Authors and Affiliations
Corresponding author
Additional information
The University of Maryland Special Research Initiative Support Program and National Institute of Health Grant R01 HL71113-01 (R.M.V.) supported this work.
Rights and permissions
About this article
Cite this article
Viscardi, R., Muhumuza, C., Rodriguez, A. et al. Inflammatory Markers in Intrauterine and Fetal Blood and Cerebrospinal Fluid Compartments Are Associated with Adverse Pulmonary and Neurologic Outcomes in Preterm Infants. Pediatr Res 55, 1009–1017 (2004). https://doi.org/10.1203/01.pdr.0000127015.60185.8a
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000127015.60185.8a
This article is cited by
-
Maternal body mass index and risk of intraventricular hemorrhage in preterm infants
Pediatric Research (2018)
-
Chemokine and cytokine levels in the lumbar cerebrospinal fluid of preterm infants with post-hemorrhagic hydrocephalus
Fluids and Barriers of the CNS (2017)
-
The Airway Microbiome at Birth
Scientific Reports (2016)
-
Role of selected cytokines in the etiopathogenesis of intraventricular hemorrhage in preterm newborns
Child's Nervous System (2016)
-
Serum eotaxin-1 is increased in extremely-low-birth-weight infants with bronchopulmonary dysplasia or death
Pediatric Research (2015)
