
Abstract
The POU1F1 gene encodes a transcription factor that is important for the development and differentiation of the cells producing GH, prolactin, and TSH in the anterior pituitary gland. Patients with POU1F1 mutations show a combined pituitary hormone deficiency with low or absent levels of GH, prolactin, and TSH. Fourteen mutations have been reported in the POU1F1 gene up to now. These genetic lesions can be inherited either in an autosomal dominant or an autosomal recessive mode. We report on the first Italian patient, a girl, affected by combined pituitary hormone deficiency. The patient was found to be positive for congenital hypothyroidism (with low TSH levels) at neonatal screening. Substitutive therapy was started, but subsequent growth was very poor, although psychomotor development was substantially normal. Hospitalized at 10 mo she showed hypotonic crises, growth retardation, delayed bone age, and facial dysmorphism. In addition to congenital hypothyroidism, GH and prolactin deficiencies were found. Mutation DNA analysis of the patient's POU1F1 gene identified the novel Q167K amino acid change at the heterozygous level. The highly conserved Q167 residue is located in the POU-specific domain. No mutation was detected in the other allele. DNA analysis in the proband's parents did not identify this amino acid substitution, suggesting a de novo genetic lesion. From these data it can be hypothesized that the Q167K mutation has a dominant negative effect.
Similar content being viewed by others


Main
CPHD is a rare inborn heterogeneous disorder. Two transcription factors, the pituitary-specific transcription factor POU1F1 (MIM 173110, usually named PIT-1) and PROP-1, the Prophet of POU1F1, which is required for POU1F1 expression (MIM 601538), may be implicated. However, several other regulatory factors, some yet unknown, play a crucial role in the pituitary hormones' action, and they can also be the cause of pituitary disease (1–7). POU1F1 acts as transactivator for GH, PRL, and β-TSH genes in the anterior pituitary gland in combination with other factors (8–11). POU1F1 recognizes the consensus binding sites within the regulation elements of the genes it controls. Deficiency of this transcription factor causes decreased levels of pituitary hormones with hypoplasia of the anterior pituitary and subsequently severe growth retardation (12). The human POU1F1 gene has been mapped to chromosome 3p11 (13), and contains six exons spanning >17 kb (14). The 1050-bp POU1F1 cDNA has been cloned (15), and it encodes a protein of 291 amino acids. This protein is characterized by two important regions: POU-S and POU-H domains, both of which are DNA binding regions. Its three-dimensional structure shows four α helices in the POU-S region and three α helices in the POU-H domain (16). The functional form of the human POU1F1 factor is an homodimer formed by interactions between the POU-S domain and the POU-H domain of two symmetrically related monomers (9, 16, 17). Dimerization occurs both by hydrophobic contacts and hydrogen binding between the two helices α3 and α4 of the POU-S domain in one monomer and the carboxyl terminus of the POU-H domain in another monomer (16, 17).
Two regulatory regions, which are responsible for transcriptional activation, autoregulation, and cell-specific expression of the human POU1F1 gene, have been identified upstream of the human POU1F1 gene, one distal (−11.8/−10.9 kb) and one proximal (−7.1/−2.3 kb). The distal regulatory region acts as a silencer, and the proximal region is a strong enhancer (18).
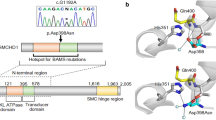
Up to now, 14 different genetic lesions—11 missense (9, 19–26), two nonsense mutations (27, 28), and one deletion (22) —have been reported in the POU1F1 gene. Figure 1 shows the schematic representation of the POU1F1 gene and the relative mutations reported in the literature according to the nomenclature of Dunnen and Antonarakis (29). They are spread throughout the gene, usually occurring in exons 4 and 6, and they are inherited either in an autosomal dominant or in an autosomal recessive mode. The most common mutation, reported in patients with CPHD, is the R271W amino acid substitution, which has been demonstrated to have a dominant negative effect. This mutation alters the POU1F1 protein, which binds normally to DNA but inhibits transcription (19). Moreover, this mutation seems to be a hot spot for POU1F1 mutations (30).

Schematic representation of the POU1F1 gene listing the new and the known mutations identified. The new mutation is indicated with an asterisk. Designation of the new and previously published POU1F1 gene mutations corresponds to the nomenclature of Dunnen and Antonarakis (29). Numbers in parentheses indicate reference citations.
We report clinical and molecular studies on the first Italian patient, a girl, affected by CPHD. Molecular analysis identified a novel mutation (Q167 K), at the heterozygous status, in the patient's POU1F1 gene.
METHODS
The study was approved by the Institutional Review Board of the Department of Pediatrics, Florence University, and informed consent was obtained from parents.
Case report.
SL, the first daughter of unrelated Italian parents, was admitted to our hospital at the age of 10 mo because of recurrent crises of hypotonia, pallor, drowsiness, and sweating. She was delivered by cesarean section at the 36th week of a first uncomplicated pregnancy. Her Apgar score was 5 and 7 at 1 and 5 min, respectively, her birth weight was 2400 g (35th centile), her length was 44 cm (15th centile), and her cranial circumference was 33 cm (60th centile). Immediately after birth cyanosis appeared and resuscitation procedures were instituted; neonatal screening led to suspected congenital hypothyroidism (Table 1), with the demonstration of a normal thyroid gland on ultrasound and no 99mTc-Pertec uptake on thyroid scintigraphy. Replacement therapy with l-thyroxine sodium (5 μg·kg−1·d−1) was started, but, in spite of this treatment, subsequent growth was very poor, although psychomotor development was substantially normal. Chromosome analysis was normal (46XX).
Clinical examination on admission showed a prominent forehead, a depressed nasal bridge, anteverted nostrils, bluish sclerae, and a small facial skull: length was 56 cm (−5.25 SD), with an evident disproportion between the US and the LS, owing to an abnormal prevalence of the former (US/LS = 2.2, normal for age 1.6); weight was 4600 g (below the 3rd percentile), and head circumference was 41.5 cm (−2 SD).
Blood and urine routine analyses were normal, but several remarkable nonketotic hypoinsulinemic hypoglycemic events (minimum 36 mg/dL) were detected: cortisol, as well as FT3 and FT4 (Table 1), was normal for the patient's age. Basal LH and FSH levels (<0.7 and 0.6 mIU/mL, respectively) were low, but the GnRH stimulation test showed normal secretion patterns of both hormones (LH peak, 6.51 mUI/mL; FSH peak, 17.9 mUI/mL).
u-TSH was low and basal PRL was undetectable; moreover, basal IGF-I and IGF-BP3 were under normal range (Table 1). Bone age was between 0 and 3 mo, according to Greulich and Pyle (30a).
At magnetic resonance imaging the anterior pituitary height was 2.5 mm, at the very lower limit of normal size (31). The signal of the posterior pituitary gland showed a normal location. EEG was normal.
hGH stimulation tests (arginine, clonidine, and growth hormone releasing factor) all showed a total unresponsiveness, and a similar result was obtained by the TSH releasing hormone test, performed to evaluate TSH and PRL secretion patterns (Table 1).
Substitutive hGH therapy (0.6 UI/d) was begun, together with an increase of daily l-thyroxine sodium dosage up to 8 μg/kg with a complete and immediate disappearance of hypoglycemic events and a normalization of IGF-I and IGF-BP3 levels (Table 1).
After 6 mo follow-up height was 71 cm (−3.6 SD, with a mean monthly gain of 2.5 cm); US/LS ratio was 1.6 (normal for age, 1.52); weight was 6850 g; bone age (12 mo, chronologic age of 18 mo) showed a remarkable recovery.
Genomic DNA analysis of the POU1F1 gene.
Genomic DNA was isolated from the patient's peripheral blood lymphocytes and from her parents' lymphocytes.
The genomic fragments covering all six exons and their flanking introns were amplified using a set of primers located in flanking intronic region sequences, (Table 2). PCR amplification was performed under the following conditions: denaturation at 94°C for 3 min followed by 30 cycles with denaturation at 94°C for 1 min, annealing temperature of 60°C for 1 min, and extension at 72°C for 1 min. The reaction mixture was carried out in a total volume of 25 μL containing 200 ng of template DNA, 2.5 U of Ampli Taq DNA polymerase (Polymed, Sambuca-Firenze, Italy), 30 pmol of each forward and reverse primer, 2.5 mM of dNTPs, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1 mM MgCl2, and 0.01% gelatin. To facilitate the amplification of exon 5, first a nested PCR with primers 4F and 6R was performed, followed by the 5F-5R amplification.
Genomic DNA analysis of the PROP-1 gene.
The three exons and intronic boundary sequences of the PROP-1 gene were amplified by PCR using primer and PCR conditions reported by Deladöey et al.(32).
DNA sequencing.
PCR products were run on a 2% agarose gel containing ethidium bromide, and DNA was visualized with an UV transilluminator. DNA fragments were excised and purified using QIAQuick Gel Extraction kit (QUIAGEN, Hilden, Germany). Approximately 100 ng of purified amplifications was used in sequencing reactions. Both strands were sequenced with the same primers used for PCR amplification. The sequencing reactions were performed using Big Dye Terminator Cycle Sequencing Ready Reaction Kit reagents (Applied Biosystems, Foster City, CA, U.S.A.). The reactions were run on an ABI 310 sequencer (Applied Biosystems) and were analyzed using Sequencing Analysis software, version 3.3. Full-length POU1F1 and PROP-1 DNAs and intron/exon boundaries were sequenced in both strands.
Restriction site analysis.
To confirm the new mutation Q167K, a modified reverse genomic primer 167MUT CTGCAGATTTTCAAATCGGCAGATTGTTGTTC, which contained the c500A>G nucleotide substitution, was used, to generate a new Taq I restriction site. Four microliters of PCR product of fragment 167MUT-4F (Table 2) was incubated for 1 h at the temperature indicated by the manufacturer's instructions, with a reaction mixture containing 2 μL of 10× reaction buffer and 5 U of Taq I restriction enzyme. The total volume was brought up to 20 μL. Digested products were analyzed on a 2% agarose gel.
RESULTS
Genomic analysis of the POU1F1 gene.
Mutation analysis of the POU1F1 gene was performed using genomic DNA sequencing and restriction enzyme in the patient, her parents, and control subjects.
The new Q167K mutation, which is caused by a C to A (c499C>A) transversion in exon 4, was found in the patient's genomic DNA. The mutation was present at the heterozygous level, and no other genetic lesion in the POU1F1 gene was detected. The direct sequence performed in the patient's parents' DNA did not show any genetic lesion in their alleles, demonstrating that this mutation has occurred de novo.
Restriction analysis using the PCR fragment carrying an artificial amplification-created restriction site was performed. The c499C>A base change destroys a Taq I restriction site introduced by PCR amplification with the modified primer 167MUT. After Taq I digestion, the normal 197-bp sequence was cut into fragments of 161 bp and 33 bp, whereas the mutant fragment remained uncut (Fig. 2). Enzymatic digestion was used to screen 200 control chromosomes from unrelated individuals, and a benign polymorphism for the changed amino acid (Q167K) was excluded.

Molecular analysis of family's DNA. A, partial nucleotide sequence of the patient's POU1F1 gene showing the new c499C>A transversion leading to Q167K amino acid substitution identified at heterozygous level. The direct sequence performed in the patient's parents' DNA did not identify any genetic lesion, showing that this mutation has occurred de novo (P, patient; M, mother; F, father). B, enzymatic restriction analysis for detection of Q167K using genomic DNA. A Taq I restriction site was generated in the wild-type PCR fragment using the 4F-167Mut primers. Lane 1, Patient; Lane 2, Mother; Lane 3, Father; Lane 4, PCR control not digested.
Genomic analysis of the PROP-1 gene.
The PROP-1 gene, which could be the cause of the CPHD phenotype, was also sequenced to exclude the possible presence of a genetic lesion responsible, in combination with the Q167K mutation in the POU1F1 gene, for the patient's phenotype.
Genomic analysis of the PROP-1 gene on the patient's DNA was performed, and all three exons of this gene were sequenced. No mutations in the entire coding sequence of the PROP-1 gene and in the exon/intron boundaries were identified. However, two known polymorphisms were identified by direct sequencing: an intronic polymorphic site located at nucleotide 109+3 of intron 1 (IVS 1+3) and a T to C transition at the nucleotide 27 position (c27 T>C), which did not introduce any amino acid substitution (A9A) (32, 33). Both polymorphisms were detected at the homozygous level.
DISCUSSION
We report a female patient in whom the diagnosis of congenital hypothyroidism was made at neonatal screening. Low values of basal T4 with a low u-TSH level led to the diagnosis of secondary hypothyroidism. Hormonal replacement therapy with l-thyroxine sodium allowed normal psychomotor development, but severe growth retardation was not sufficiently taken into consideration. She was referred to our clinic at the age of 10 mo, after a first episode of hypotonia, pallor, drowsiness, and sweating. During hospitalization, other hypotonic crises with concomitant hypoglycemia occurred. These episodes, as well as growth failure and low TSH levels, led us to suspect CPHD, which was confirmed by the endocrinologic evaluations (Table 1). Among reported CPHD patients central hypothyroidism varies in severity and age of onset. However, in many cases hypothyroidism was found before GH deficiency (34).
The size of the pituitary gland at magnetic resonance imaging was at the lower limit of the normal range. In CPHD patients, the size of the pituitary can vary enormously from small to normal to enlarged (4, 35, 36). In the POUF1 defect anterior pituitary size is generally small or normal (36). However, it has also been hypothesized that the hypoplasia of the pituitary gland could be a late-onset and age-dependent manifestation (37),.
Several transcriptional factors (POU1F1, PROP-1, LHX3, PTX2, HESX1, and so forth) are involved in the development of the pituitary gland and in the proliferation of pituitary cell populations (9). A deficiency in one of these factors leads to the CPHD phenotype. Most molecular studies carried out in patients affected by CPHD with GH, PRL, and TSH deficiencies have shown genetic lesions in the PROP-1 gene (20, 32, 36), even if Brown et al.(38) reported that approximately half of CPHD cases can be attributed to mutations in the POU1F1 gene.
To clarify a possible molecular defect leading to CPHD in this patient, the POU1F1 gene was sequenced. A novel missense mutation, Q167K, of the POU1F1 gene at the heterozygous level was identified. The mutation was not detected in the patient's unaffected parents.
Comparison with other proteins of the POU family in several species has shown that the Q167 amino acid is a highly conserved residue (Fig. 3). This amino acid is the first residue of the third α helix (α3) of the POU-S domain. In particular, the three residues, Q167, T168, and R173, lying within helix α3 of the POU-S domain, form hydrogen bonds with DNA base pairs (16). Crystallographic analyses have shown that this helix α3 has the most contacts with the major grooves of DNA in the consensus binding sites of the target genes (16, 17). The Q167K causes a change in polarity. A neutral glutamine residue is changed with a basic lysine residue, and the decreased hydrophobicity could affect the correct protein folding.

Alignment of a POU protein fragment with similar domain. hPIT-1 (POU1F1 human), rPIT-1 (rat), mPIT-1 (mouse), Oct-1 and Oct-2 (human transcription factors), and Unc-86 (POU protein of the Caenorhabditis elegans). Amino acids that are highly conserved are boxed.
In our patient, despite the sequencing of the entire coding region and the exon/intron boundaries, the only Q167K genetic lesion was detected at the heterozygous level.
Because only one mutation at the heterozygous status was identified in the POU1F1 gene, and taking into account that mutations in the human PROP-1 gene lead to a form of CPHD with the same symptomatology before the age of puberty (39), the patient's PROP-1 gene was analyzed. Direct sequencing of the PROP-1 gene did not show any genetic alteration associated with the patient's CPHD phenotype. The PROP-1 gene is required for POU1F1 activation, and its deficiency involves the production of GH, PRL, and TSH hormones and later of FSH and LH. According to the molecular data, the patient's gonadotropin secretions, in spite of low basal levels, were normal, as demonstrated by the gonadotropin releasing hormone stimulative test. Mutations in the POU1F1 gene, reported in the literature, can cause CPHD of both autosomal recessive and dominant inheritance. Up to now, 14 different genetic lesions have been reported in the POU1F1 gene, and four of them (K216E, R271W, P14L, and P24L) have been shown to be inherited as a dominant trait (9, 19–21). The pathogenic mechanism of dominant inheritance has been extensively explored for only two mutations, R271W and K216E (9, 19). R271W occurs near the end of the POU-H domain. This mutant protein shows a greater affinity for DNA binding sites of the target genes than the wild-type protein. Therefore, the dominant mechanism, probably caused by competitive inhibition of the mutant homodimer and/or the mutant/wild type heterodimer as opposed to the wild-type homodimer, leads to a block in transcription (19). The other well-characterized dominant effect has been studied for the K216E mutation (9). The K216E mutant protein acts on the distal POU1F1 gene promoter, which contains the retinoic receptor binding site and the POU1F1 protein binding site. The latter is necessary for autoregulation at negative feedback. The activation of transcription needs both factors as well as retinoic acids. The K216E mutant protein is unable to form a complex with retinoic acid and its receptor, causing a block of the POU1F1 gene transcription. The decreased levels of POU1F1 mRNA and subsequently of the POU1F1 protein give rise to a CPHD phenotype (9).
Based on these data and considering that a dominant mechanism can occur in this gene, we hypothesize that the Q167K amino acid substitution in the POU1F1 gene causes a dominant molecular defect. This defect can be responsible for a POU1F1 expression reduction by inhibition of the POU1F1 autoregulation or a decrease in DNA binding affinity. This genetic lesion must be considered pathogenic as it was not detected in 200 normal chromosomes. Besides, the Q167K involves a highly conserved amino acid (Fig. 3), located within the important third α helix of the POU-S domain. The absence of this mutation in the patient's parents supports the hypothesis of a de novo mutation with a dominant negative effect.
Nevertheless, a second genetic lesion outside the POU1F1 coding sequence or intron/exon boundaries tested (promoter, enhancer, and so forth) or a deletion larger than one of the sequenced fragments cannot be excluded. Moreover, a combined effect by an alteration in another correlated gene, even if it has never been reported in CPHD cases, could be possible.
CONCLUSIONS
In conclusion, we report a patient affected by CPHD, in whom central hypothyroidism was detected at neonatal screening thanks to simultaneous TSH and T4 determination. This allowed early replacement therapy, with a subsequent normal psychomotor development. Nevertheless, GH deficiency caused hypoglycemic crises and severe growth retardation. Further biochemical and molecular investigations led to a CPHD diagnosis owing to a defect of the transcriptional factor POU1F1 with a possible dominant effect. In patients with central congenital hypothyroidism, attention should be paid to linear growth to avoid a late diagnosis of GH deficiency. Moreover, a GH and ACTH evaluation is advisable because hypoglycemia and subsequent brain damage may occur in young children before failure to thrive.
Abbreviations
- CPHD:
-
combined pituitary hormone deficiency
- PRL:
-
prolactin
- FT4:
-
free thyroxine
- T4:
-
total thyroxine
- u-TSH:
-
ultra thyroid-stimulating-hormone
- US:
-
upper segment
- LS:
-
lower segment
- IGF-BP3:
-
insulin growth factor-binding protein 3
- FT3:
-
free triiodothyronine
- POU-S:
-
POU-specific
- POU-D:
-
POU-homeo
REFERENCES
Howard PW, Maurer RA 1994 Thyrotropin releasing hormone stimulates transient phosphorylation of the tissue-specific transcription factor, Pit-1. J Biol Chem 269: 28662–28669
Hamilton J, Chitayat D, Blaser S, Cohen LE, Phillips JA, Daneman D 1998 Familial growth hormone deficiency associated with MRI abnormalities. Am J Med Genet 80: 128–132
Lipkin SM, Naar AM, Kalla KA, Sack RA, Rosenfeld MG 1993 Identification of a novel zinc finger protein binding a conserved element critical for Pit-1-dependent growth hormone gene expression. Genes Dev 7: 1674–1687
Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers AV, Goossens M, Gruters A, Amselem S 2000 Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet 25: 182–186
Sloop KW, Parker GE, Hanna KR, Wright HA, Rhodes SJ 2001 LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene 265: 61–69
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC 1998 Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet 19: 125–133
Machinis K, Pantel J, Netchine I, Leger J, Camand OJ, Sobrier ML, Dastot-Le Moal F, Duquesnoy P, Abitbol M, Czernichow P, Amselem S 2001 Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet 69: 961–968
Ingraham HA, Chen RP, Mangalam HJ, Elsholtz HP, Flynn SE, Lin CR, Simmons DM, Swanson L, Rosenfeld MG 1988 A tissue-specific transcription factor containing a homeodomain specifies a pituitary phenotype. Cell 55: 519–529
Cohen LE, Zanger K, Brue T, Wondisford FE, Radovick S 1999 Defective retinoic acid regulation of the Pit-1 gene enhancer: a novel mechanism of combined pituitary hormone deficiency. Mol Endocrinol 13: 476–484
Bradford AP, Brodsky KS, Diamond SE, Kuhn LC, Liu Y, Gutierrez-Hartmann A 2000 The Pit-1 homeodomain and beta-domain interact with Ets-1 and modulate synergistic activation of the rat prolactin promoter. J Biol Chem 275: 3100–3106
Simmons DM, Voss JW, Ingraham HA, Holloway JM, Broide RS, Rosenfeld MG, Swanson LW 1990 Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev 4: 695–711
Pfaffle R, Kim C, Otten B, Wit JM, Eiholzer U, Heimann G, Parks J 1996 Pit-1: clinical aspects. Horm Res 45: 25–28
Raskin S, Cogan JD, Summar ML, Moreno A, Krishnamani MR, Phillips JA 1996 Genetic mapping of the human pituitary-specific transcriptional factor gene and its analysis in familial panhypopituitary dwarfism. Hum Genet 98: 703–705
Ohta K, Nobukuni Y, Mitsubuchi H, Ohta T, Tohma T, Jinno Y, Endo F, Matsuda I 1992 Characterization of the gene encoding human pituitary-specific transcription factor, Pit-1. Gene 122: 387–388
Lew AM, Elsholtz HP 1991 Cloning of the human cDNA for transcription factor Pit-1. Nucleic Acids Res 19: 6329
Jacobson EM, Li P, Leon-del-Rio A, Rosenfeld MG, Aggarwal AK 1997 Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev 11: 198–212
Scully KM, Jacobson EM, Jepsen K, Lunyak V, Viadiu H, Carriere C, Rose DW, Hooshmand F, Aggarwal AK, Rosenfeld MG 2000 Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science 290: 1127–1131
Rajas F, Delhase M, De La Hoya M, Verdood P, Castrillo JL, Hooghe-Peters EL 1998 Nuclear factor 1 regulates the distal silencer of the human PIT1/GHF1 gene. Biochem J 333: 77–84
Radovick S, Nations M, Du Y, Berg LA, Weintraub BD, Wondisford FE 1992 A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency. Science 257: 1115–1118
Fofanova OV, Takamura N, Kinoshita E, Yoshimoto M, Tsuji Y, Peterkova VA, Evgrafov OV, Dedov II, Goncharov NP, Yamashita S 1998 Rarity of PIT1 involvement in children from Russia with combined pituitary hormone deficiency. Am J Med Genet 77: 360–365
Ohta K, Nobukuni Y, Mitsubuchi H, Fujimoto S, Matsuo N, Inagaki H, Endo F, Matsuda I 1992 Mutations in the Pit-1 gene in children with combined pituitary hormone deficiency. Biochem Biophys Res Commun 189: 851–855
Pellegrini-Bouiller I, Belicar P, Barlier A, Gunz G, Charvet JP, Jaquet P, Brue T, Vialettes B, Enjalbert A 1996 A new mutation of the gene encoding the transcription factor Pit-1 is responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 81: 2790–2796
Pfaffle RW, DiMattia GE, Parks JS, Brown MR, Wit JM, Jansen M, Van der Nat H, Van den Brande JL, Rosenfeld MG, Ingraham HA 1992 Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science 257: 1118–1121
Brown MR, Parks JS, Adess ME, Rich BH, Rosenthal IM, Voss TC, VanderHeyden TC, Hurley DL 1998 Central hypothyroidism reveals compound heterozygous mutations in the Pit-1 gene. Horm Res 49: 98–102
Gat-Yablonski G, Lazar L, Pertzelan A, Phillip M 2002 A novel mutation in PIT-1: phenotypic variability in familial combined pituitary hormone deficiencies. J Pediatr Endocrinol Metab 15: 325–330
Pernasetti F, Milner RD, al Ashwal AA, de Zegher F, Chavez VM, Muller M, Martial JA 1998 Pro239Ser: a novel recessive mutation of the Pit-1 gene in seven Middle Eastern children with growth hormone, prolactin, and thyrotropin deficiency. Clin Endocrinol Metab 83: 2079–2083
Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, Kohno H 1992 Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet 1: 56–58
Irie Y, Tatsumi K, Ogawa M, Kamijo T, Preeyasombat C, Suprasongsin C, Amino N 1995 A novel E250X mutation of the PIT1 gene in a patient with combined pituitary hormone deficiency. Endocr J 42: 351–354
Dunnen JT, Antonarakis SE 2000 Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15: 7–12
Cohen LE, Wondisford FE, Salvatoni A, Maghnie M, Brucker-Davis F, Weintraub BD, Radovick S 1995 A “hot spot” in the Pit-1 gene responsible for combined pituitary hormone deficiency: clinical and molecular correlates. J Clin Endocrinol Metab 80: 679–684
Greulich WW, Pyle SI 1959 Radiographic Atlas Skeletal Development of the Hand and Wrist. 2nd ed. Standford University Press, Stanford, CA.
Argyropoulou M, Perignon F, Brauner R, Brunelle F 1992 Magnetic resonance imaging in the diagnosis of growth hormone deficiency. J Pediatr 120: 886–891
Deladoey J, Fluck C, Buyukgebiz A, Kuhlmann BV, Eble A, Hindmarsh PC, Wu W, Mullis PE 1999 “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 84: 1645–1650
Duquesnoy P, Roy A, Dastot F, Ghali I, Teinturier C, Netchine I, Cacheux V, Hafez M, Salah N, Chaussain JL, Goossens M, Bougneres P, Amselem S 1998 Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency. FEBS Lett 437: 216–220
Holl RW, Pfaffle R, Kim C, Sorgo W, Teller WM, Heimann G 1997 Combined pituitary deficiencies of growth hormone, thyroid stimulating hormone and prolactin due to Pit-1 gene mutation: a case report. Eur J Pediatr 156: 835–837
Mendonca BB, Osorio MG, Latronico AC, Estefan V, Lo LS, Arnhold IJ 1999 Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301, G302 in the PROP1 gene. J Clin Endocrinol Metab 84: 942–945
Parks JS, Brown MR 1999 Transcription factors regulating pituitary development. Growth Horm IGF Res 9: 2–11
Ward L, Chavez M, Huot C, Lecocq P, Collu R, Decarie JC, Martial JA, Van Vliet G 1998 Severe congenital hypopituitarism with low prolactin levels and age-dependent anterior pituitary hypoplasia: a clue to a PIT-1 mutation. J Pediatr 132: 1036–1038
Brown MR, Parks JS, Adess ME, Rich BH, Rosenthal IM, Voss TC, VanderHeyden TC, Hurley DL 1998 Central hypothyroidism reveals compound heterozygous mutations in the Pit-1 gene. Horm Res 49: 98–102
Pfaffle RW, Blankenstein O, Wuller S, Kentrup H 1999 Combined pituitary hormone deficiency: role of Pit-1 and Prop-1. Acta Paediatr Suppl 88: 33–41
Acknowledgements
The authors thank Dr. M. Cappellini for providing the patient's pituitary gland size.
Author information
Authors and Affiliations
Corresponding author
Additional information
Partially supported by funds from Cassa di Risparmio di Firenze, Fondi di Ateneo (Ex MURST 60%).
Rights and permissions
About this article
Cite this article
Malvagia, S., Poggi, G., Pasquini, E. et al. The De Novo Q167K Mutation in the POU1F1 Gene Leads to Combined Pituitary Hormone Deficiency in an Italian Patient. Pediatr Res 54, 635–640 (2003). https://doi.org/10.1203/01.PDR.0000084113.41375.1E
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000084113.41375.1E
This article is cited by
-
POU1F1 mutations in combined pituitary hormone deficiency: differing spectrum of mutations in a Western-Indian cohort and systematic analysis of world literature
Pituitary (2021)
-
Association of pituitary specific transcription factor-1 (POU1F1) gene polymorphism with growth and biometric traits and blood metabolites in Iranian Zel and Lori-Bakhtiari sheep
Molecular Biology Reports (2014)
-
Associations of POU1F1 gene polymorphisms and protein structure changes with growth traits and blood metabolites in two Iranian sheep breeds
Journal of Genetics (2014)
-
Analysis of polymorphism within POU1F1 gene in relation to milk production traits in dairy Sarda sheep breed
Molecular Biology Reports (2012)
-
A novel recessive splicing mutation in the POU1F1 gene causing combined pituitary hormone deficiency
Journal of Endocrinological Investigation (2009)
