
Abstract
Degradation of troponin I (TnI) by calpain occurs with myocardial stunning in ischemia-reperfusion injury. Glucocorticoids attenuate myocardial ischemia-reperfusion injury, but their effect on TnI degradation is unknown. A piglet model was used to test the hypotheses that cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA) are associated with TnI degradation and that TnI alterations could be prevented by glucocorticoid treatment. Piglets were cooled to 18°C, subjected to 2 h of circulatory arrest, rewarmed to 37°C, and allowed to recover for 2 h. Methylprednisolone was administered 6 h before surgery (3 0 mg/kg) and at initiation of CPB (30 mg/kg). The untreated group received saline. Left ventricular tissue was collected after recovery and analyzed by Western blot for TnI, calpain, and calpastatin (the natural inhibitor of calpain). CPB/DHCA animals had 27.4 ± 0.2% of total detected TnI present in degraded form. Glucocorticoid treatment significantly decreased the percentage of degraded TnI (12.0 ± 0.1%, p < 0.05). Calpain I and calpain II increased after CPB/DHCA compared with non-CPB/DHCA controls (p < 0.05), with or without glucocorticoid treatment. Calpastatin significantly decreased in untreated CPB/DHCA animals compared with non-CPB/DHCA controls (p < 0.05), but levels were preserved by glucocorticoids. Glucocorticoids were associated with preservation of maximum rate of increase of left ventricular pressure at 95 ± 10% of baseline, whereas maximum rate of increase of left ventricular pressure decreased to 62 ± 12% of baseline without steroids. TnI degradation occurs after CPB/DHCA in neonatal pigs. Reduction in reperfusion injury by glucocorticoids may depend partly on preservation of calpastatin activity and intact TnI.
Similar content being viewed by others


Main
Surgery for congenital heart disease often requires CPB and periods of myocardial ischemia. A period of total circulatory arrest may also be required, particularly for complex neonatal repairs. In the first 8 to 12 h after CPB there is a transient decrease in myocardial function consistent with reperfusion injury (1). Although the exact mechanisms of CPB-related myocardial injury remain unknown, there is substantial evidence that CPB can produce a systemic inflammatory response or free radical injury (2–4). The cysteine protease calpain exists in the myocardium in two isoforms, calpain I and calpain II. Recently, it has been suggested that activation of calpain during reperfusion of ischemic myocardium may be a key event in the process of myocardial stunning (5, 6). Calcium influx that occurs when ischemic myocardium is reperfused promotes dissociation of calpain from its inhibitor, calpastatin. Activated calpain then degrades the contractile regulatory protein TnI by cleavage at the C-terminus to form the fragment TnI(1-193). Additional systematic degradation of the amino terminus can lead to subsequent smaller degradation products. Degradation products and covalent complexes of TnI have been observed in humans with ischemic heart disease (7, 8).
The use of glucocorticoids either before or during CPB can lead to improved cardiopulmonary function after open-heart surgery (9, 10). Although myocardial stunning has many similarities to the type of myocardial reperfusion injury that occurs after CPB and circulatory arrest, the effect of steroids on calpain, calpastatin, and TnI degradation in the context of CPB-related myocardial injury has not been studied. Rather, the main focus of research on the effects of steroids on post-CPB myocardial function has been on the ability of steroids to reduce generation of proinflammatory cytokines (11, 12). As TnI degradation by calpain can be observed in vitro (5) in the absence of white blood cells, it is possible that myocardial dysfunction after CPB may not be entirely dependent on a significant inflammatory response. Steroids may, in fact, directly affect the calpain-calpastatin system, because glucocorticoids can preserve calpastatin activity in healthy skeletal muscle despite inducing a decrease in calpastatin mRNA levels (13).
Much of the work on the calpain-calpastatin system in large animal models has focused on ischemia in fully mature myocardium, analogous to human coronary artery disease. It is well established that the neonatal myocardium handles calcium differently than the adult myocardium, and that neonates (<1 mo old) are at particularly high risk for myocardial dysfunction after CPB. We therefore chose to examine CPB with DHCA in 2-wk-old piglets. Piglets of this age have routinely been used to mimic the neonatal human heart (14, 15). This model was designed to simulate congenital heart surgery in the context of the immature myocardium. We tested the hypotheses that CPB/DHCA is associated with TnI degradation and that steroids before and during CPB prevent this phenomenon. To better understand the mechanisms by which steroids might prevent TnI degradation induced by CPB/DHCA, we examined calpain and calpastatin in treated and untreated hearts along with the associated changes in hemodynamics after CPB/DHCA. The significant findings are that CPB/DHCA is associated with TnI degradation, increased calpain, and decreased calpastatin. Steroid administration improved hemodynamics and lessened TnI degradation. This appears to be related to preservation of calpastatin.
METHODS
Animal protocol.
All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” prepared by the Institute of Laboratory Animals Resources and published by the National Institutes of Health (National Institutes of Health publication no. 86-23, revised 1985). The Animal Care and Use Committee at Children's Hospital Research Foundation also approved the protocol.
Two-week-old crossbred piglets weighing 4-7 kg were divided into four groups. Group 1 (CPB/DHCA/steroid, n = 6) received methylprednisolone (30 mg/kg, intramuscularly) 6 h before CPB and again in the CPB pump priming solution (30 mg/kg). Group 2 (CPB/DHCA, n = 6) underwent CPB/DHCA but received only sterile saline in volumes similar to the glucocorticoid-treated animals. Groups 3 and 4 were non-CPB controls. Group 3 (no CPB/DHCA, n-6) underwent sternotomy and immediate removal of LV tissue. Sterile saline, volume equal to the steroid-treated groups, was administered to these animals 6 h before sternotomy. Group 4 (no CPB/DHCA/steroid, n = 6) were given methylprednisolone (30 mg/kg) 6 h before sternotomy and LV tissue collection. This steroid dosing regimen is similar to that used by others to blunt the inflammatory response to CPB (12).
The animals were anesthetized with ketamine (22 mg/kg, intramuscularly) and acepromazine (1.1 mg/kg, intramuscularly), intubated, and mechanically ventilated. Continuous pentobarbital infusion (20 mg·kg−1·h−1), intermittent fentanyl citrate (10 μg·kg−1·h−1), and pancuronium bromide (0.1 mg·kg−1·h−1) were used throughout the experimental protocol. Median sternotomy exposed the heart, and pressure catheters (Millar Instruments, Houston, TX, U.S.A.) were placed in the LV. A Doppler flow probe (Transonics Systems, Inc, Ithaca, NY, U.S.A.) was placed around the pulmonary artery to measure cardiac output. Baseline measurements of pulmonary artery and systemic arterial pressures, cardiac output, LV end-diastolic pressure, and LV dP/dt were taken after a 30-min equilibration period. Oxygen saturation, pH, hematocrit, electrolytes, and lactate were determined using a blood gas analyzer (Bayer Diagnostics, Emeryville, CA, U.S.A.).
Heparin (300 U/kg) was given, and CPB was initiated via the carotid artery and right atrium. The CPB priming solution consisted of 800 mL of direct-drawn whole porcine blood (Animal Biotech Industries, Danboro, PA, U.S.A.) stabilized with sodium citrate. Animals were cooled to a rectal temperature of 18°C during 40 min to ensure even and thorough cooling. For DHCA the bypass circuit was turned off and the head packed in ice. The heart was protected with topical cold saline and ice, with the pericardial well maintained below 10°C. DHCA was maintained for 120 min, after which CPB was reinstituted and the animals were rewarmed to 37°C during 40 min. Piglets were removed from CPB and maintained for 2 h, during which time cardiopulmonary function was monitored continuously. Transmural LV biopsies from the same area of the outer ventricular wall in each of the animals were obtained at the terminal time point.
Tissue specimens.
LV myocardium for immunoblotting of total tissue was flash-frozen in liquid nitrogen and homogenized in 10 mM 3-(N-morpholino)propanesulfonic acid (100 mg/mL). Homogenates were centrifuged at 4°C at 21,000 × g, and the supernatant was retained. The protein concentration for each sample was determined by Bio-Rad protein assay (Bio-Rad, Hercules, CA, U.S.A.). The samples were frozen at −80°C until used.
Cardiac myofibrils for immunoblotting of TnI were prepared according to Solaro et al. (16) Myofibrils, rather than total tissue, were used for the blots because of the high concentration of TnI present in myofibrils. Briefly, tissue samples were homogenized in standard buffer containing 60 mM KCl, 30 mM imidazole, 2 mM magnesium chloride, and EDTA-free protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A.). Homogenates were centrifuged at 4°C at 21,000 × g, the pellets were resuspended, then pelleted two more times. The final pellet was homogenized in buffer containing 1 mM EGTA. Samples were again centrifuged at 4°C at 21,000 × g, and the pellets were resuspended in buffer containing 1% Triton X-100, homogenized, and centrifuged. Triton X-100 was washed from the myofibrils by four washings with buffer. Purified myofibrils were dissolved in buffer to a concentration of 0.5 mg/mL. Concentrated myofibrils were stored at −80°C until assayed.
Western blots.
For TnI, 5 μg of purified cardiac myofibrils were separated on 12% Novex Bis-Tris precast gels (Invitrogen, Carlsbad, CA, U.S.A.) using the method described by Laemmli (17). Calpain and calpastatin immunoblots were separated on 10% Bis-Tris gels with 20 μg of protein per well. After electrophoresis the proteins were transferred to polyvinylidene fluoride membranes (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.). Molecular weight markers (Invitrogen) were used to identify protein bands of known size.
TnI was identified with a polyclonal antibody to human cardiac TnI (RDI-TROPIabg; Research Diagnostics, Inc, Flanders, NJ, U.S.A.). This antibody is specific for cardiac TnI and is not reactive with human slow skeletal muscle TnI. Blots were also probed with the TnI MAb 8I-7 (Spectral Diagnostics Inc, Whitestone, VA, U.S.A.). The total protein blots were used to detect levels of calpain I, calpain II, and calpastatin with commercially available antibodies (Calbiochem, San Diego, CA, U.S.A.). A GAPDH antibody (MAB374; Chemicon International, Temecula, CA, U.S.A.) was used to compare lanes for equal loading. Blots used for quantitative analyses were loaded so that intact TnI, the densest band, was in the linear range of the analytical software. Primary antibody concentrations were 1:500 for the polyclonal TnI, 1:2500 for the monoclonal TnI, 1:5000 for calpastatin, 1:1000 for calpain I, 1:500 for calpain II, and 1:5000 for GAPDH. Incubation was overnight at 4°C. The blots were rinsed with Tris-buffered saline with Triton-X (TBST), and horseradish peroxidaseconjugated secondary antibodies were added and incubated for 1 h at 25°C. A chemiluminescence Western blotting system (Roche Molecular Biochemicals) detected proteins of interest. The density of each band was analyzed with the NIH Image software package (National Institutes of Health, Bethesda, MD, U.S.A.). The density of calpain and calpastatin was expressed as a ratio to GAPDH in the same lane to correct for differences in loading. The density of each TnI band was expressed as a percentage of all TnI detected in the same lane.
Cardiomyocyte culture.
Because porcine cardiomyocytes grow poorly in culture, neonatal rat cardiomyocytes were used for this part of the experiment and were isolated as previously described (18). Briefly, hearts were removed from 1- to 3-d-old rats and digested with collagenase (Worthington Biochemical Co, Lakewood, NJ, U.S.A.) in PBS. After preplating, cardiomyocyte cultures contained less than 5% contaminating cell types. Cells were maintained in 5% carbon dioxide in Dulbecco's Modified Eagle Medium with 2% fetal bovine serum, penicillin, and streptomycin for 48 h. Cells were then moved to media containing 10% fetal bovine serum, and some cultures had methylprednisolone added at different concentrations. Six hours later some cultures had 1 U/mL calpain I (Calbiochem) and 10 mM calcium chloride added to the media. Incubation was continued for 24 h at which time the cells were scraped from the plate, myofibrils were isolated, and Western blot analyses for TnI were performed as described above.
Statistical analyses.
Repeated-measures ANOVA was used to compare hemodynamic values across all time points, followed by Fisher's least significant difference post hoc test when appropriate. Differences between treatments at the same time point were analyzed with single-factor ANOVA, as were differences in protein levels. A p ≤ 0.05 was considered significant (StatView 4.01, Abacus Concepts Inc, Berkeley, CA, U.S.A.). Data are expressed as the mean ± SD.
RESULTS
TnI degradation.
Analysis of LV myofibrils for TnI degradation identified intact TnI at 29 kD as well as bands representing degradation products at 26 kD and 15 kD in both of the CPB/DHCA groups (groups 1 and 2, Fig. 1). There were also higher molecular weight compounds of 65 kD and 53 kD, which presumably were degraded and covalently complexed TnI fragments. Minimal TnI degradation was identified in the untreated or steroid-treated control animals not subjected to CPB/DHCA (groups 3 and 4). Animals exposed to CPB/DHCA who did not receive steroids (group 2) had a significantly greater percentage of total detected TnI present in either degraded or covalently complexed form compared with steroid-treated animals (group 1; 27.4 ± 6% versus 12.0 ± 3%; p = 0.05; Fig. 1). The greatest difference between steroidtreated and untreated animals was in the percent of total TnI present as the 26-kD degradation product that represents TnI(1-193). There was also a tendency toward a greater percentage of total detectable TnI present as the 15-kD degradation product in untreated animals. There were no differences between treated and untreated animals in the percentage of TnI present as covalent complexes. Similar Western blots probed with the 8I-7 monoclonal anti-TnI antibody failed to demonstrate differences in degradation product size or abundance compared with those identified with the cardiac TnI-specific polyclonal antibody despite the more C-terminal epitope of the polyclonal antibody.

A, representative Western blot probed with anti-TnI antibody showing intact TnI at 29 kD, 26-kD and 15-kD degradation products, and larger covalent complexes. Lanes 1-4, CPB/DHCA. Lanes 5-8, no CPB/DHCA. Animals in lanes 2, 4, 6, and 8 received methylprednisolone 6 h before CPB (30 mg/kg) and at the initiation of CPB (30 mg/kg). Animals in lanes 1, 3, 5, and 7 received saline. B, histogram showing the percentage of all detected TnI present as degraded TnI (all degradation products and covalent complexes combined) and as the 26-kD and 15-kD degradation products and covalent complexes. *p < 0.05 vs non-CPB/DHCA controls, ‡p < 0.05 vs CPB/DHCA without steroids.
Cardiomyocyte culture.
To determine whether steroids could decrease calpain-mediated TnI degradation in vitro, cultured neonatal rat cardiomyocytes were treated with exogenous calpain I with or without pretreatment with steroids. As expected, myocytes not pretreated with steroids showed degradation of TnI as evidenced by the appearance of 26-kD and 15-kD degradation products (Fig. 2). Calpain-treated cardiomyocytes showed less degradation of TnI after steroid administration in what appeared to be a dose-dependent fashion.

Western blot of myofibrils from cultured neonatal rat cardiomyocytes probed with anti-TnI antibody. Intact TnI at 29 kD and the 26-kD and 15-kD degradation products are identified. Cells were treated as follows. Lane 1: no calpain, no methylprednisolone; Lane 2: calpain I, no steroids; Lane 3: calpain I, 31 μg/mL methylprednisolone; Lane 4: calpain I, 156 μg/L methylprednisolone; Lane 5: calpain I, 312 μg/mL methylprednisolone.
Calpain levels.
We measured protein levels of both calpain I and calpain II in each of the treatment groups because differences in TnI degradation between steroid-treated and untreated animals might be the result of increased levels of calpain in untreated myocardium. Both calpain I and calpain II were significantly elevated in the myocardium of animals that underwent CPB/DHCA compared with control animals that did not undergo bypass and circulatory arrest (Fig. 3). There were no differences in the levels of calpain I and II between steroidtreated and untreated animals in either non-CPB/DHCA controls or in the CPB/DHCA groups.

A, representative Western blot probed with antibodies for calpain I or II and GAPDH. Lanes 1-4, CPB/DHCA. Lanes 5-8, no CPB/DHCA. Animals in lanes 3, 4, 7, and 8 received methylprednisolone 6 h before CPB (30 mg/kg) and at the initiation of CPB (30 mg/kg). Animals in lanes 1, 2, 5, and 6 received saline. B, histogram showing calpain I and II levels as a ratio to GAPDH. *p < 0.05 vs no CPB/DHCA.
Calpastatin levels.
Calpastatin, the endogenous inhibitor of calpain, could be responsible for the preservation of TnI in steroid-treated animals. Calpastatin in the LV of CPB/DHCA animals that did not receive steroids was significantly decreased compared with controls, but steroid treatment of CPB/DHCA animals preserved calpastatin at a level comparable to the groups not undergoing CPB/DHCA (p < 0.005). Steroids alone, in the absence of CPB, did not affect calpastatin levels as evidenced by the lack of difference between the groups not undergoing CPB/DHCA (Fig. 4).

A, representative Western blot probed with anti-calpastatin and anti-GAPDH. Lanes 1-4, CPB/DHCA. Lanes 5-8, no CPB/DHCA. Animals in lanes 3, 4, 7, and 8 received methylprednisolone 6 h before CPB (30 mg/kg) and at the initiation of CPB (30 mg/kg). Animals in lanes 1, 2, 5, and 6 received saline. B, histogram showing calpastatin as a ratio of GAPDH. *p < 0.05 vs no CPB/DHCA and steroid-treated CPB/DHCA.
Hemodynamics.
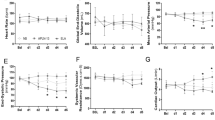
In the CPB/DHCA group LV + dP/dt fell to 67% of baseline at 90 min and to 62% of baseline at 120 min of recovery (p < 0.05; Fig. 5). Administration of glucocorticoids before and during CPB maintained LV +dP/dt at 99% and 95% of baseline at 90 and 120 min of recovery, respectively. Although cardiac output was lower in both groups at 120 min of recovery (p < 0.05, data not shown), animals receiving glucocorticoid therapy maintained their cardiac output longer into the experiment and at a higher level than CPB/DHCA animals at each time. Systemic vascular resistance in control and glucocorticoid-treated animals did not change from baseline during the recovery period.

LV +dP/dt was significantly decreased at 90 and 120 min after CPB/DHCA in untreated animals (·). Systolic function was largely maintained in steroid-treated animals (▪). *p < 0.05 vs CPB/DHCA/steroid.
DISCUSSION
The important findings in this study are that 120 min of CPB/DHCA is associated with TnI degradation in the immature porcine heart and that this is ameliorated by glucocorticoid treatment. Glucocorticoids preserve myocardial calpastatin levels after CPB/DHCA, which may offset concomitant increases in calpain. The protective effect of steroids was observed even in isolated cardiomyocytes treated with exogenous calpain, suggesting this is a direct effect of steroids not mediated through antiinflammatory mechanisms. Steroid-treated animals also have improved hemodynamics.
Although TnI modification has clearly been demonstrated in rats subject to ischemia and reperfusion (5) and in the serum of humans after myocardial infarction (7), it has been more difficult to detect in porcine models of stunned myocardium. Thomas et al. (19) reported that adult pigs with clinical evidence of stunning after either 10 min of total ischemia or 60 min of partial ischemia had no increase in TnI(1-193), but TnI(1-193) did become apparent after 1-2 h of ischemia. Similarly Kim and colleagues (20), also using adult pigs, failed to demonstrate evidence of TnI degradation in a model of stunning using 90 min of coronary stenosis followed by 60 min of reperfusion. Myocardial dysfunction in this model was attributed to alterations in calcium handling and phospholamban phosphorylation. Despite these findings, the work by Kim et al. (20) did demonstrate that porcine myocardium is capable of producing TnI(1-193) in response to exogenously administered calpain. Species-specific differences in either antibody sensitivity or mechanisms responsible for myocardial stunning have been speculated to account for the discrepancies between studies in porcine hearts and other animals (21). Our results clearly demonstrate TnI degradation with CPB/DHCA, 2 h of global ischemia, and subsequent reperfusion. Furthermore, the differences between steroid-treated and control animals were observed in samples run together and then processed on a single gel using an antibody specific for cardiac TnI. Use of the monoclonal anti-TnI antibody 8I-7 showed the same degradation products.
Our experiments differ in several important ways from the above studies. The current study used immature piglets and CPB/DHCA because we are specifically interested in the type of myocardial injury seen in newborns and infants after congenital heart surgery. This is an important distinction because maturational differences in cardiomyocyte structure and function could alter the effects of CPB and surgical ischemiareperfusion. TnI degradation by calpain is ultimately a calcium-dependent process. Inasmuch as neonatal hearts may handle calcium differently than adult hearts (22) and because the proteins that make up the troponin complex, including TnI, may undergo developmental alteration (23, 24), examination of this type of injury in the context of the immature heart is essential. The contention that immature hearts are more susceptible to reperfusion injury after CPB is consistent with data from Imura and colleagues (25), who demonstrated increased release of TnI into the bloodstream after CPB and cardioplegia in infants <1 y old compared with older children. Furthermore, 3-d-old porcine hearts are more susceptible to calcium-mediated reperfusion injury than are adult hearts (26), a finding consistent with the present study. As with the human heart, an exact timeline for the progression of cardiomyocyte maturation has not been clearly established in a porcine model. The 2-wk-old piglets used in this study are intended to be a model of the neonatal human heart, although, as with the human heart, the exact ultrastructural developmental stage is unknown. Nevertheless, similar porcine models have been used by other investigators to study neonatal myocardium (14, 15). Overall, our results support the contention that the immature heart may be uniquely sensitive to the ischemia and reperfusion that occur with congenital heart surgery.
Our model of ischemia also differs significantly from those of other investigators. Both ischemic and reperfusion times (2 h each) were longer than those reported in studies that previously failed to demonstrate TnI degradation in porcine myocardium. Although Thomas and colleagues (19) did show that TnI(1-193) is present in the porcine myocardium after ischemic periods of >1 h, this appeared to be associated with ischemic injury rather than with the reversibility that characterizes stunned myocardium. We cannot exclude the possibility that the contractile dysfunction in our experiments was caused by irreversible injury rather than transient myofilament damage. Clinical studies of cardiac dysfunction after CPB/DHCA in human neonates demonstrates the time-dependent decrease in cardiac output and reversibility that characterize myocardial stunning (1), but our experiments were terminated at 2 h after reperfusion, before reversibility could be formally demonstrated. Similarly, it is not possible to distinguish between the effects of CPB, myocardial ischemia, or DHCA alone. Our experiments were designed to mimic clinical conditions that occur during repair of congenital heart disease rather than to be a precise model of stunning or to isolate the exact aspect of cardiac surgery that is most responsible for myocardial injury. The observation that TnI degradation is associated with such injury and that this phenomenon is prevented by steroids is by itself clinically important. Further support for the applicability of this model of myocardial injury is that modification of cardiac TnI occurs in human adults after CPB (8). Although no clearly defined pattern of TnI modification has yet emerged, the data suggest that such modification can occur. The present study is the first to show extensive TnI degradation specifically in association with CPB/DHCA.
The finding that calpastatin levels are reduced after CPB and that this reduction is prevented by steroid pretreatment is likewise unique. Although the mechanisms responsible were not examined in the present work, other studies have suggested that calpastatin may be preserved by steroids. Exposure of healthy skeletal muscle to dexamethasone results in decreased mRNA expression for both calpain and calpastatin, but activity of both is maintained (13). These findings, together with our data, suggest that steroids may prevent loss of calpastatin, although mRNA levels were not examined in the current study and thus increased transcription cannot be ruled out. Increased transcription might be suggested by the observation that the calpastatin promoter region is known to contain a cAMP response element. Calpastatin might therefore be up-regulated by steroids secondary to preservation of the β-adrenergic receptor response (27). The calpastatin promoter has not been documented to be steroid responsive.
One limitation of this study is that we cannot prove conclusively that the increased calpastatin and decreased TnI degradation we observed are directly responsible for the preservation of ventricular function in the steroid-treated animals. Steroids may in fact have other important effects in the immature heart and after CPB/DHCA, including increasing L-type calcium current (28, 29) or through antiinflammatory properties (9, 10). Nevertheless, there is significant evidence that calpain activation and TnI degradation are sufficient to cause myocardial dysfunction. Myocardial trabeculae exposed to calpain I in vitro exhibit decreased calcium responsiveness characteristic of stunned myocardium (5). Excised, arrested rat hearts subject to 12 h of cold ischemia in the presence of a calpain inhibitor retain myocardial function better than hearts not treated with inhibitor (30). Similarly, the calpain inhibitor calpeptin can block TnI degradation and preserve ventricular function in preload-injured isolated rat hearts (31). Transgenic mice that overexpress TnI(1-193) develop a cardiomyopathy, strongly suggesting that TnI degradation is sufficient to induce myocardial dysfunction (6). Preservation of intact TnI by glucocorticoids after CPB/DHCA is thus likely to be of hemodynamic benefit.
CONCLUSIONS
In summary, use of glucocorticosteroids before and during CPB/DHCA in immature piglets is associated with improved systolic LV function, decreased TnI degradation, and increased myocardial calpastatin. TnI preservation in steroid-treated animals occurred despite similar increases in calpain in treated and untreated animals. In vitro experiments using cultured cardiomyocytes suggest that inhibition of TnI degradation by steroids is mediated through direct effects and not secondary to their antiinflammatory properties. Although further understanding of the multiple effects steroids may have in the neonatal cardiovascular and immune systems is needed, it appears their actions may be more complex than previously appreciated.
Abbreviations
- CPB:
-
cardiopulmonary bypass
- DHCA:
-
deep hypothermic circulatory arrest
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- LV:
-
left ventricle
- TnI:
-
troponin I
- LV dP/dt:
-
maximum rate of increase of left ventricular pressure
REFERENCES
Wernovsky G, Wypij D, Jonas RA, Mayer JE Jr, Hanley FL, Hickey PR, Walsh AZ, Chang AC, Castaneda AR, Newburger JW, Wessel DL 1995 Postoperative course and hemodynamic profile after the arterial switch operation in neonates and infants: a comparison of low-flow cardiopulmonary bypass and circulatory arrest. Circulation 92: 2226–2235
Wan S, LeClerc JL, Vincent JL 1997 Cytokine responses to cardiopulmonary bypass: lessons learned from cardiac transplantation. Ann Thorac Surg 63: 269–276
Hall RI, Smith MS, Rocker G 1997 The systemic inflammatory response to cardiopulmonary bypass: pathophysiological, therapeutic, and pharmacological considerations. Anesth Analg 85: 766–782
Kilbridge PM, Mayer JE, Newburger JW, Hickey PR, Walsh AZ, Neufeld EJ 1994 Induction of intercellular adhesion molecule-1 and E-selectin mRNA in heart and skeletal muscle of pediatric patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg 107: 1183–1192
Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E 1997 Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res 80: 393–399
Murphy AM, Kogler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marban E 2000 Transgenic mouse model of stunned myocardium. Science 287: 488–491
Labugger R, Organ L, Collier C, Atar D, Van Eyk JE 2000 Extensive troponin I, and T modification detected in serum from patients with acute myocardial infarction. Circulation 102: 1221–1226
McDonough JL, Labugger R, Pickett W, Tse MY, MacKenzie S, Pang SC, Atar D, Ropchan G, Van Eyk JE 2001 Cardiac troponin I is modified in the myocardium of bypass patients. Circulation 103: 58–64
Jansen NJ, van Oeveren W, van den Broek L, Oudemans-van Straaten HM, Stoutenbeek CP, Joen MC, Roozendaal KJ, Eysman L, Wildevuur CR 1991 Inhibition by dexamethasone of the reperfusion phenomena in cardiopulmonary bypass. J Thorac Cardiovasc Surg 102: 515–525
Tabardel Y, Duchateau J, Schmartz D, Marecaux G, Shahla M, Barvais L, Leclerc JL, Vincent JL 1996 Corticosteroids increase blood interleukin-10 levels during cardiopulmonary bypass in men. Surgery 119: 76–80
Volk T, Schmutzler M, Engelhardt L, Docke WD, Volk HD, Konertz W, Kox WJ 2001 Influence of aminosteroid and glucocorticoid treatment on inflammation and immune function during cardiopulmonary bypass. Crit Care Med 29: 2137–2142
Lodge AJ, Chai PJ, Daggett CW, Ungerleider RM, Jaggers J 1999 Methylprednisolone reduces the inflammatory response to cardiopulmonary bypass in neonatal piglets: timing of dose is important. J Thorac Cardiovasc Surg 117: 515–522
Yeh JY, Ou BR, Forsberg NE 1994 Effects of dexamethasone on muscle protein homeostasis and on calpain and calpastatin activities and gene expression in rabbits. J Endocrinol 141: 209–217
Brix-Christensen V, Tonnesen E, Hjortdal VE, Chew M, Flo C, Marqversen J, Hansen JF, Andersen NT, Ravn HB 2002 Neutrophils and platelets accumulate in the heart, lungs, and kidneys after cardiopulmonary bypass in neonatal pigs. Crit Care Med 30: 670–676
Kronon MT, Allen BS, Halldorsson A, Rahman S, Barth MJ, Ilbawi M 2002 Delivery of a non-potassium modified maintenance solution to enhance myocardial protection in stressed neonatal hearts: a new approach. J Thorac Cardiovasc Surg 123: 119–129
Solaro RJ, Pang DC, Briggs FN 1971 The purification of cardiac myofibrils with Triton X-100. Biochim Biophys Acta 245: 259–262
Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685
Lim HW, New L, Han J, Molkentin JD 2001 Calcineurin enhances MAPK phosphatase-1 expression and p38 MAPK inactivation in cardiac myocytes. J Biol Chem 276: 15913–15919
Thomas SA, Fallavollita JA, Lee TC, Feng J, Canty JM Jr 1999 Absence of troponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine. Circ Res 85: 446–456
Kim SJ, Kudej RK, Yatani A, Kim YK, Takagi G, Honda R, Colantonio DA, Van Eyk JE, Vatner DE, Rasmusson RL, Vatner SF 2001 A novel mechanism for myocardial stunning involving impaired Ca2+ handling. Circ Res 89: 831–837
Foster DB, Van Eyk JE 1999 In search of the proteins that cause myocardial stunning. Circ Res 85: 470–472
Hohl CM, Livingston B, Hensley J, Altschuld RA 1997 Calcium handling by sarcoplasmic reticulum of neonatal swine cardiac myocytes. Am J Physiol 273:H192–H199
Anderson PA, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK 1995 Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ Res 76: 681–686
Hunkeler NM, Kullman J, Murphy AM 1991 Troponin I isoform expression in human heart. Circ Res 69: 1409–1414
Imura H, Caputo M, Parry A, Pawade A, Angelini GD, Suleiman MS 2001 Agedependent and hypoxia-related differences in myocardial protection during pediatric open heart surgery. Circulation 103: 1551–1556
Wittnich C, Wallen WJ, Belanger MP, Ikonomidis JS 1999 Extracellular calcium concentration affects susceptibility to global ischemic injury in newborn but not adult hearts. J Heart Lung Transplant 18: 675–683
Cong M, Thompson VF, Goll DE, Antin PB 1998 The bovine calpastatin gene promoter and a new N-terminal region of the protein are targets for cAMP-dependent protein kinase activity. J Biol Chem 273: 660–666
Whitehurst RM Jr, Zhang M, Bhattacharjee A, Li M 1999 Dexamethasone-induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca2+ current. J Mol Cell Cardiol 31: 1551–1558
Wang L, Feng ZP, Duff HJ 1999 Glucocorticoid regulation of cardiac K+ currents and L-type Ca2+ current in neonatal mice. Circ Res 85: 168–173
Saito T, Mishima A, Asano M, Ukai T, Yamamoto S, Kunimatsu M, Sasaki M, Manabe T 1999 Protective effects of calpain inhibitor for prolonged hypothermic cardiac preservation. Jpn J Thorac Cardiovasc Surg 47: 145–152
Feng J, Schaus BJ, Fallavollita JA, Lee T, Canty JM 2001 Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103: 2035–2037
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by The Children's Heart Foundation, Chicago, IL (J.M.P.) and by National Institutes of Health HD22827 (S.M.S.).
Rights and permissions
About this article
Cite this article
Schwartz, S., Duffy, J., Pearl, J. et al. Glucocorticoids Preserve Calpastatin and Troponin I during Cardiopulmonary Bypass in Immature Pigs. Pediatr Res 54, 91–97 (2003). https://doi.org/10.1203/01.PDR.0000065730.79610.7D
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000065730.79610.7D
This article is cited by
-
Corticosteroid effects on ventilator-induced diaphragm dysfunction in anesthetized rats depend on the dose administered
Respiratory Research (2010)
-
Effect of dexamethasone on postoperative cardiac troponin T production in pediatric cardiac surgery
Intensive Care Medicine (2005)
