
Abstract
Obesity is a complex disease that involves interactions between environmental and genetic factors. Obesity results from an imbalance between food intake and energy expenditure over several years. The genetic approach both in animal models and in humans has allowed immense progress in the understanding of body weight regulation. Monogenic forms of obesity in humans have been characterized and result from mutations in genes involved in the central pathways of food intake regulation. However, these cases are extremely rare and generally obesity must be considered as a complex polygenic disease involving interactions between multiple genes and the environment. Numerous studies, including studies in children, have tried to identify “susceptibility” genes. At present, the results are not conclusive inasmuch as they are highly variable between studies and because the relative risk associating a specific gene allele and obesity remains low. Thus, it seems highly premature to genotype obese patients on a large scale for predictive testing. When specific pharmacological treatments based on recent discoveries become available, however, genetic testing could help discriminate between the subtypes of obesity that may respond differentially to treatment.
Similar content being viewed by others

Main
Obesity has become a major health problem in modern societies, with a prevalence of up to 25% in certain countries and an increasing incidence in children (1). Concurrently, the genetic approach of obesity is progressing rapidly. This brief review discusses the mechanisms of energy storage, the relationship between obesity and genetics, and the question of the place of genetics in obesity treatment.
ENERGY BALANCE
If energy expenditure (mechanical work and heat) is smaller than the intake, energy will be stored. The major form of energy storage is triglyceride contained in adipose tissue. The storage of energy is a physiologic process involved in survival. Indeed, the release of fatty acids by the hydrolysis of white adipose tissue triglyceride stores allows us to face periods of food shortage or increased energy expenses.
White adipocytes originate from precursor cells through a commitment and differentiation process that can take place during the whole lifespan (2). Adipose tissue localization is different in males (typically the upper part of the trunk and intra-abdominal) and females (the lower part of the trunk and subcutaneous). Intra-abdominal obesity is more frequently associated with cardiovascular disease (3).
Another key function of adipose tissue is the secretion of factors, including cytokines, angiogenic factors, immunorelated factors, prostaglandins, angiotensinogen, and proteins involved in the regulation of energy balance and carbohydrate metabolism (e.g. resistin, adiponectin) (4). Some of these factors are adiposity signals secreted in proportion to the adipose tissue mass and could be involved in the development of the obesity-related complications.
Energy balance can be regulated either at the level of food intake or energy expenditure. Considerable progress in the understanding of energy storage regulation stems from the discovery of leptin. Leptin is a cytokine-like polypeptide produced by the adipocyte that controls food intake through the activation of hypothalamic receptors (5). Leptin is produced proportionally to the adipose mass and thus informs the brain of the fat store level. In the hypothalamic arcuate nucleus, leptin induces the synthesis and secretion of α-MSH from the prohormone POMC. α-MSH binds to the MC4R in hypothalamic nuclei and inhibits food intake. Leptin also decreases the expression of orexic peptides such as neuropeptide Y. Other hormones, ghrelin (orexic), insulin (anorexic), and cholecystokinin (anorexic), are involved in the short-term control of energy intake.
Furthermore, in rodents, leptin can regulate energy output by stimulating sympathetic activation of brown adipose tissue (2). This tissue is characterized by the presence of a specific β-adrenergic receptor (β3) and a high number of mitochondria. In the inner membrane of these mitochondria, UCP1 allows the production of heat from fatty acid oxidation. Thus, when this tissue is activated, energy oversupply can be dissipated as heat, thus reducing fat storage. Brown adipose tissue exists in small (rodents) or young mammals, including the human baby. It persists through adulthood in rodents but tends to disappear in adult humans. Brown adipose tissue has been described in human adults only in the vicinity of adrenal tumors (pheochromocytomas) and in some groups exposed for very long periods to cold. Other UCP-like proteins (2 and 3) have been described in various tissues, including muscles, but their role in thermogenesis is considered as doubtful. Thus, it is yet unclear whether mechanisms exist to regulate energy output in humans.
OBESITY
Obesity is defined as an accumulation of excess body fat, to such an extent that health might be impaired (World Health Organization definition). In their daily practice, physicians use a BMI (body weight in kilograms divided by the square of height in meters) >30 to define obesity (even though BMI is not an accurate reflection of the adipose mass). BMI charts are also available for children and take into account age and sex (6). Different types of obesity (android, gynoid) can be defined according to the location of adipose tissue depots. Usually, obesity involves an increase in both the number and size of adipocytes (2). In vivo studies in humans, including children and adolescents, have demonstrated that the development of obesity over time is mostly the result of periods of overfeeding rather than a defect in basal energy expenditure (7).
ENVIRONMENTAL AND GENETIC FACTORS
As stated above, energy storage in the form of fat is an important adaptation for survival. Thus, it is likely that combination of genes have been selected during evolution to favor energy storage (the “thrifty gene” hypothesis) (2). In our context of increased food availability and decreased physical activity, these genes will confer a susceptibility to the development of obesity and its maintenance (8). The involvement of genetic factors in the control of body weight is indicated by studies of monozygotic twins showing a high concordance of body composition and response to overfeeding (8). The genetic susceptibility is in most cases polygenic, with each gene probably contributing a small part, and is rarely the result of a Mendelian gene (monogenic obesity). Whether or not a smaller number of genes with larger phenotypic effects (i.e. major genes) will be discovered in common obese population is still unknown.
In addition to the fact that genetic factors can modulate nutrient storage, nutrients are able to modulate gene expression (9). Thus, obesity results from complex genetic and environmental interactions. This renders the search for susceptibility genes in humans extremely difficult.
GENETIC SYNDROMES CONCOMITANT WITH OBESITY
Obesity is one of the features accompanying numerous genetic syndromes (at least 25). Most of the more common syndromic forms of obesity, like Prader-Willi, Cohen, Alstrom, and Bardet-Biedl (BBS), have been genetically mapped (10). The challenge is now to determine the specific genes responsible—some were recently identified, such as MMKS in BBS or ALM1 in Alstrom disease—and the proteins they encode. Finally, it will be necessary to determine their role as susceptibility genes for obesity, diabetes, sensory deficit, and more generally in diseases with multiple and heterogeneous features.
HUMAN MONOGENIC OBESITY WITH KNOWN GENE AND PROTEIN FUNCTION
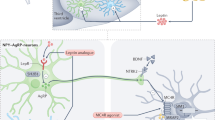
The first gene identification in obese human subjects was linked with the screening for genes identified previously in rodent models of monogenic obesity. These rodents models of obesity mostly involved genes in the regulatory pathway of food intake. Similar affected pathways have been characterized in human monogenic obesity (Fig. 1).

Schematic representation of leptin regulation of food intake and proteins mutated in monogenic human obesities. Leptin is secreted by adipocytes. It binds to hypothalamic receptors in the arcuate nucleus and this induces, among other effects, an increased synthesis and secretion of α-MSH. α-MSH is formed from POMC through proteolytic cleavage mediated by PC-1. α-MSH binds to the MC4R in the paraventricular nucleus. This, in turn, inhibits the effectors of food intake. The steps that have been identified as mutated in human monogenic forms of obesity are circled with a dotted line.
Leptin and leptin receptor.
Homozygous carriers of a loss of function mutation in the leptin gene exhibit morbid obesity with onset in the first months of life, hypogonadotropic hypogonadism and central hypothyroidism (8). Affected subjects continuously seek food and eat considerably more than their siblings. None of the heterozygous parents or siblings are morbidly obese. In one case, a leptin-deficient child has been treated by leptin replacement. In this 9-y-old girl, daily s.c. injection of recombinant human leptin for a year was well tolerated and led to an important and sustained fat mass loss and an age-appropriate improvement in function of the reproductive axis (8).
One family has been identified with a leptin receptor mutation (11). In the three subjects with the homozygous mutation, a truncation of the receptor abolishes leptin signaling, leading to a phenotype similar to that of individuals with leptin deficiency, although more severe. The three sisters bearing the leptin receptor mutation also display a significant growth retardation caused by impaired GH secretion. At present, seven cases have been described for leptin or leptin receptor mutations (10).
Melanocortin pathway.
POMC is a peptide expressed in human brain, gut, placenta, and pancreas. It is the precursor of many hormones, including ACTH and α-MSH produced by PC-1-dependent cleavage (5). Because α-MSH is involved in the regulation of food intake and also in hair pigmentation, it could be predicted that the phenotype associated with a defect in POMC function would include obesity, altered pigmentation, and ACTH deficiency. As expected, two children with homozygous or compound heterozygous loss-of-function mutations in POMC exhibited a phenotype including obesity, red hair, and adrenal insufficiency, reflecting the lack of pituitary neuropeptides derived from the POMC gene (12). Mutations of PC-1 have also been described and are associated with obesity and ACTH insufficiency, as for POMC mutations. However, these subjects also demonstrate hyperproinsulinemia, because PC-1 is involved in the conversion of proinsulin into insulin in the pancreatic β-cell (8).
Contrasting with the forms of monogenic obesity associated with multiple endocrine dysfunctions, mutations in the MC4R (the receptor of α-MSH) cause dominant and recessive inherited nonsyndromic obesity with incomplete penetrance (the mutation is not always associated with obesity) and variable expression (a similar mutation can yield various degrees of obesity) (8, 13). Human obesity caused by MC4R mutations is similar to more common forms of obesity, with an earlier age of onset. Interestingly, a trend toward a greater incidence of childhood obesity and an excessive hunger and food-seeking behavior from the age of 6 to 8 mo have been reported. Additionally, the common association of pediatric obesity with increased growth velocity was also noted. MC4R mutations represent a significant cause of obesity in morbidly obese children and adults (0.5–6%) (14). At least 27 different mutations in 68 individuals have been described (10). The role of these mutations remains to be clarified inasmuch as individuals with these various mutations have different levels of obesity.
Even considering this latter case, however, obesity in which a single gene can be identified as the major cause is rare. Most of the genes involved in monogenic forms of obesity are involved in the regulation of food intake. However, this does not preclude that in the most common forms of obesity (see below) genes involved in numerous pathways, from nutrient absorption to energy expenditure play a role.
COMMON FORMS OF OBESITY
Susceptibility genes.
Two approaches are possible to identify susceptibility genes. The first one consists of genome-wide scans aimed at detecting chromosomal regions showing linkage with obesity in large collections of nuclear families, mostly comprising adult sibling pairs. Fifty-nine loci have been linked to obesity across all chromosomes except chromosome Y, but no gene involved in common obesity has been characterized yet (10). The second method is the candidate gene approach. It involves testing the association between obesity and a specific allele of a gene that appears to be a good candidate (e.g. a gene involved in the regulation of food intake), either in a family study or in large cohorts of unrelated controls and patients.
Candidate genes: regulation of food intake.
Plasma leptin in obese subjects is usually normal for their fat mass, indicating that leptin deficiency is not a primary cause of common obesity. However, linkage and association studies have produced evidence, although sometimes inconsistent, for the involvement of leptin—or at least leptin gene locus—in the complex determinism of obesity and its related phenotypes in early onset obesity. One gene variant, adjacent to sequences involved in the leptin gene transcription, was shown to modulate the response to caloric restriction in severely obese women. Homozygous obese girls carrying the variant allele had a 25% decrease in leptin levels despite a similar body fat mass (15). Similar relationships were described between a variant in the exon 1 noncoding region and circulating leptin levels in French obese adults. These studies suggest that genetic variation in the leptin gene may account for the variation in circulating leptin levels and thus could modify the ability of the brain to sense the amount of fat stored in white adipose tissue. Contrasting to genetic studies on the leptin gene, no association was confirmed between obesity and the leptin-receptor gene in a meta-analysis (16).
Although still limited in number and importance compared with adult studies, other genes have been evaluated as plausible candidates in children and in populations with a prevalence of moderate to severe juvenile obesity. Some of these linkage or association studies in young obese population are described by Rankinen et al(10). The POMC gene is a plausible candidate for polygenic obesity. It is present in one locus and linked with obesity in genome-wide scan analysis. One study describes higher leptin levels in Italian obese children with a mutation in POMC gene (17). Functional studies as well as analysis in larger populations are needed to confirm the involvement of POMC in the development of common obesity in children.
Other neuropeptides involved in the control of food intake such as CART (cocaine- and amphetamine-regulated transcript), or neuromedin B (a bombesin-related peptide) have also been tested as plausible candidates but inconclusive results were found.
Candidate genes: regulation of metabolism.
Insulin is considered an important adiposity signal for the brain. In a study of early onset obesity, the effect of variants in the insulin gene among 615 obese children were analyzed. Interestingly, it was shown that a specific allelic combination induces a higher insulin secretion and a higher risk of developing juvenile obesity (18).
Candidate genes also include genes involved in pathways of energy expenditure and lipid and adipose tissue metabolism. Beta-adrenergic receptors (β2 and β3) as well as the UCP1 have been the target of many association studies, including studies in children. However, these studies have yielded, on the whole, inconclusive results. A study performed in adolescent girls suggests that carriers of a specific variant of PPARγ, a transcription factor involved in adipocyte differentiation and metabolism (2), have an increased annual rate of change of BMI (19). In contrast, in adult patients, association studies with variants of PPARγ yielded limited or uncertain associations with obesity phenotypes. Thus, in such a multifactorial and polygenic disease, it is at present extremely difficult to associate a gene with a higher risk for obesity in a clear and reproducible manner.
Obese children represent an interesting target population for genetic studies because the variation of their body fat mass and the occurrence of obesity might be less subject to environmental pressure. However, to date, the increasing number of association studies in children indicate that the role of candidate genes in early onset obesity may be modest and should be considered in combination with other factors, exactly as in adult populations. Accuracy of the genome-wide scan approach in its capacity to identify the causative genes has still to be demonstrated.
WHAT CAN PEDIATRICIANS LEARN FROM GENETIC STUDIES IN OBESITY?
It is unclear at the present stage whether counseling and genetic testing (predictive genetics) will be integrated into the practice of obesity prevention and management. Before this can occur, answers to several questions are needed, in addition to a response to the more general psychological, social, and ethical questions that the prospect of genetic testing can raise.
First, are we able to define the predictive risk related to obesity for gene variations or mutations in candidate genes? Although the genetic prediction for monogenic diseases is very high and can be calculated, the predictive risk is usually small and presently difficult to determine for more common causes of obesity. For example, the relative risks of having high weight gain, occurrence of diabetes, or obesity in allelic carriers of several candidate genes (β3AR, sulfonylurea receptor, apoB) are usually <5. We face here more probability than predictive medicine.
Second, are we able to specifically treat or prevent obesity in carriers of allelic variants or mutations conferring a defined risk? Currently, there is no specific and efficient drug treatment for obesity. Conventional therapy in children associates nutritional education, increased exercise, and sometimes behavioral modification and psychological and social support. Excessive pressure (like that possibly exerted on patients at risk) inexorably leads to weight regain and worsening of obesity. Is it then reasonable to target a specific population—particularly children—for prevention?
In patients with MC4R mutations, the frequency and relative risk for obesity (100-fold higher) could lead us to consider mutation screening at a population scale. MC4R agonists are now under development and might be used in the future in patients with decreased melanocortinergic activity. However, as stated above, the expression of the disease in mutation carriers is variable and the penetrance incomplete, emphasizing the role of environment as well as other genetic contributors. In addition, the described mutations have various functional consequences and their precise evaluation is still required. With all these uncertainties, it seems premature to propose the detection of the mutation on a large scale.
CONCLUSION
One of the major interests of the genetic approach, and more generally of clinical genetic studies in patients, is to provide breakthroughs in the understanding of molecular mechanisms involved in body weight regulation (8). This, in turn, could provide new targets for the development of common obesity therapies, a goal that will be reached in the near future. Predictive genetics is problematic in common forms of obesity and does not justify large-scale testing as long as specific treatments are not available. However, once these therapies are developed, genetic testing could allow classification of patients in various subgroups for which the efficacy of different treatments could be tested (8).
Abbreviations
- BMI:
-
body mass index
- MC4R:
-
melanocortin 4 receptor
- α-MSH:
-
α-melanocyte stimulating hormone
- PC-1:
-
pro-hormone convertase 1
- POMC:
-
pro-opiomelanocortin
- PPARγ:
-
peroxisome proliferator-activated receptor γ
- UCP1:
-
uncoupling protein 1
References
Troiano RP, Flegal KM 1998 Overweight children and adolescents: description, epidemiology, and demographics. Pediatrics 101: 497–504
Spiegelman BM, Flier JS 2001 Obesity and the regulation of energy balance. Cell 104: 531–543
Franco C, Bengtsson BA, Johannsson G 2001 Visceral obesity and the role of the somatotropic axis in the development of metabolic complications. Growth Horm IGF Res 11: S97–S102
Trayhurn P, Beattie JH 2001 Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60: 329–339
Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG 2000 Central nervous system control of food intake. Nature 404: 661–671
Flegal KM, Wei R, Ogden C 2002 Weight-for-stature compared with body mass index-for-age growth charts for the United States from the Centers for Disease Control and Prevention. Am J Clin Nutr 75: 761–766
Jequier E, Tappy L 1999 Regulation of body weight in humans. Physiol Rev 79: 451–480
Barsh GS, Farooqi IS, O'Rahilly S 2000 Genetics of body-weight regulation. Nature 404: 644–651
Foufelle F, Ferré P 2002 New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor SREBP-1c. Biochem J 366: 377–391
Rankinen T, Perusse L, Weisnagel SJ, Snyder EE, Chagnon YC, Bouchard C 2002 The human obesity gene map: the 2001 update. Obes Res 10: 196–243
Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B 1998 A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392: 398–401
Krude H, Gruters A 2000 Implications of proopiomelanocortin (POMC) mutations in humans: the POMC deficiency syndrome. Trends Endocrinol Metab 11: 15–22
Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P 2000 Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 106: 253–262
Dubern B, Clement K, Pelloux V, Froguel P, Girardet JP, Guy-Grand B, Tounian P 2001 Mutational analysis of melanocortin-4 receptor, agouti-related protein, and alpha-melanocyte-stimulating hormone genes in severely obese children. J Pediatr 139: 204–209
Le Stunff C, Le Bihan C, Schork NJ, Bougneres P 2000 A common promoter variant of the leptin gene is associated with changes in the relationship between serum leptin and fat mass in obese girls. Diabetes 49: 2196–2200
Heo M, Leibel RL, Fontaine KR, Boyer BB, Chung WK, Koulu M, Karvonen MK, Pesonen U, Rissanen A, Laakso M, Uusitupa MI, Chagnon Y, Bouchard C, Donohoue PA, Burns TL, Shuldiner AR, Silver K, Andersen RE, Pedersen O, Echwald S, Sorensen TI, Behn P, Permutt MA, Jacobs KB, Elston RC, Hoffman DJ, Gropp E, Allison DB 2002 A meta-analytic investigation of linkage and association of common leptin receptor (LEPR) polymorphisms with body mass index and waist circumference. Int J Obes Relat Metab Disord 26: 640–646
Miraglia del Giudice E, Cirillo G, Santoro N, D'Urso L, Carbone MT, Di Toro R, Perrone L 2001 Molecular screening of the proopiomelanocortin (POMC) gene in Italian obese children: report of three new mutations. Int J Obes Relat Metab Disord 25: 61–67
Le Stunff C, Fallin D, Schork NJ, Bougnères P 2000 The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet 26: 444–446
Witchel SF, White C, Siegel ME, Aston CE 2001 Inconsistent effects of the proline12 –> alanine variant of the peroxisome proliferator-activated receptor-gamma2 gene on body mass index in children and adolescent girls. Fertil Steril 76: 741–747
Acknowledgements
The authors thank Professor A. Basdevant for helpful discussions and Dr. B. Hegarty for kindly correcting the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clément, K., Ferré, P. Genetics and the Pathophysiology of Obesity. Pediatr Res 53, 721–725 (2003). https://doi.org/10.1203/01.PDR.0000059753.61905.58
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000059753.61905.58
This article is cited by
-
An exposome-wide association study on body mass index in adolescents using the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and 2013–2014 data
Scientific Reports (2022)
-
Sodium intake assessed by 24-h urine excretion and its relationship with anthropometric measurements in Malaysian adults
Journal of Health, Population and Nutrition (2021)
-
Genetics of Obesity
Indian Journal of Clinical Biochemistry (2016)
-
Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective
Molecular Genetics and Genomics (2015)
-
Determinants of overweight and obesity among Bangladeshi diabetic women of reproductive age
BMC Research Notes (2014)
