
Abstract
Infant macrosomia is a classic feature of a gestational diabetes mellitus (GDM) pregnancy and is associated with increased risk of adult obesity and type II diabetes mellitus, however mechanisms linking GDM and later disease remain poorly understood. The heterozygous leptin receptor-deficient (Leprdb/+) mouse develops spontaneous GDM and the fetuses display characteristics similar to infants of GDM mothers. We examined the effects of GDM on maternal insulin resistance, fetal growth, and postnatal development of hepatic insulin resistance. Fetal body weight on d 18 of gestation was 6.5% greater (p < 0.05) in pups from ad libitum-fed db/+ mothers compared with wild-type (WT) controls. Pair-feeding db/+ mothers to the intake of WT mothers normalized fetal weight despite less than normal maternal insulin sensitivity. More stringent caloric restriction reduced insulin and glucose levels below WT controls and resulted in fetal intrauterine growth restriction. The level of hepatic insulin receptor protein was decreased by 28% to 31% in both intrauterine growth restriction and fetuses from ad libitum-fed GDM mothers compared with offspring from WT mothers. In 24-wk-old adult offspring from GDM mothers, body weight was similar to WT offspring, however, the females from GDM mothers were fatter and hyperinsulinemic compared with offspring from WT mothers. Insulin-stimulated phosphorylation of Akt, a key intermediate in insulin signaling, was severely decreased in the livers of adult GDM offspring. Hepatic glucose-6-phosphatase activity was also inappropriately increased in the adult offspring from GDM mothers. These results suggest that spontaneous GDM in the pregnant Leprdb/+ mouse is triggered by overfeeding, and this effect results in obesity and insulin resistance in the livers of the adult offspring. The specific decrease in Akt phosphorylation in livers of adult offspring suggests that this may be a mechanism for reduced insulin-dependent physiologic events, such as suppression of hepatic glucose production, a defect associated with susceptibility to type II diabetes mellitus.
Similar content being viewed by others


Main
Fetal growth and development are primarily determined by the fetal genome, however, the genetic regulation of fetal growth is influenced by different maternal factors, which can exert a stimulatory or an inhibitory effect. Epidemiologic studies have revealed strong statistical links between nutritional experiences during pregnancy and later development of diseases such as obesity and type II diabetes in adulthood (1, 2). Most convincing are the studies in Pima Indians demonstrating that, besides a genetic transmission of diabetes, the diabetic intrauterine milieu can also induce a diabetogenic tendency in the offspring. IGT and obesity are two to three times more frequent in children of mothers who had diabetes during pregnancy than in children from mothers who developed diabetes after pregnancy (3). Central to this hypothesis is the fact that programming of fetal metabolism usually occurs during a critical period in prenatal or neonatal life, and that the effects persist into adulthood.
GDM is characterized by glucose intolerance first recognized during pregnancy, and is associated with fetal macrosomia, which contributes to the increased maternal and perinatal morbidity (4, 5). There is general agreement that diet is the first-line treatment for GDM, however caloric restriction during pregnancy remains controversial. Caloric restriction can improve glucose tolerance but may be associated with increased ketogenesis during pregnancy. Rizzo et al.(6) reported that the children's mental development index scores at 3–5 y of age were negatively correlated with maternal third-trimester plasma β hydroxybutyrate and FFA levels. In addition, maternal ketonuria is associated with oligohydramnios and changes in fetal heart rate (7).
A number of animal models have been developed to investigate mechanisms underlying dietary-environment triggers involved in GDM and obesity in offspring of diabetic mothers. Most of these models have been artificially derived by either eliminating maternal insulin production by streptozotocin injection or by giving large amounts of glucose or insulin to the mother or fetus. Fetuses of some of these models (notably the streptozotocin-treated rat with profound maternal and fetal hyperglycemia) are actually small for gestational age rather than macrosomic (8). In addition, most women diagnosed with GDM have IGT, usually during the second and third trimester, associated with moderate to severe insulin resistance. It is therefore not clear that these models reflect the typical metabolic abnormalities that accompany human GDM. A genetic model of GDM is the heterozygous C57BL/KsJ-Leprdb/+ mouse that develops diabetes only during pregnancy resulting from a mutation of the gene for the leptin receptor. The homozygous db/db female mouse is infertile, but the pregnant heterozygous female db/+ mouse develops IGT and elevated HbA1c, and the offspring have increased insulin and are significantly heavier than offspring from C57BL/KsJ-+/+ controls, regardless of fetal genotype (9–11).
Insulin resistance is caused by postreceptor defect(s) in the intracellular insulin signaling cascade, leading to IGT, and failure of insulin to suppress hepatic glucose production (12). The initial events include insulin binding to its receptor, activation of intracellular receptor autophosphorylation and kinase activity, and the phosphorylation of IRS-1 and IRS-2. The phosphorylation of IRS results in recruitment and tyrosine phosphorylation of the p85α regulatory subunit of PI 3-kinase (13), thereby increasing activity of the PI 3-kinase enzyme complex (14). Current evidence indicates that IRS-2-mediated PI 3-kinase activity is necessary for insulin suppression of hepatic glucose production and down-regulation of the mRNA coding for gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (15–17). The insulin-signaling pathways downstream from PI 3-kinase are beginning to be understood. Recently, protein kinase B (PKB or Akt) has been shown to function as an acute regulator of hepatic glucose output in response to insulin (18). Akt is serine phosphorylated by insulin, which leads to the down-regulation of gluconeogenic gene expression (19, 20).
Both low birth weight and fetal overgrowth are associated with insulin resistance in adulthood (21, 22), however, the mechanisms are complex and not well understood. The purpose of this study was to evaluate the effect of GDM and caloric restriction on maternal and fetal hepatic insulin resistance, and to determine how GDM impacts adult insulin resistance. Our data suggest that GDM and fetal macrosomia in this model is triggered by over-feeding. The wild-type fetuses of GDM mothers are macrosomic and demonstrate a reduction in hepatic insulin receptor protein. The adult offspring are obese and demonstrate decreased insulin-stimulated phosphorylation at the level of Akt, which may in turn be responsible for hyperinsulinemia, increased glucose-6-phosphatase, and hepatic insulin resistance.
METHODS
Animals and experimental protocol.
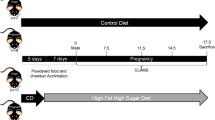
Male and female C57BL/KsJ+/+ and C57BL/KsJdb/+ mice were obtained from Jackson Laboratories (Bar Harbor, ME, U.S.A) at 6 wk of age. Mice were housed in groups of four in a temperature-, humidity-, and light-controlled (lights on at 0600 h, off at 1800 h) room. Mice were given ad libitum access to commercial mouse chow (8664 Harlan Teklad F6 Rodent Diet, Harlan Teklad, Madison, WI, U.S.A.) and water. The diet composition consisted of 29% protein, 47% carbohydrate, and 17% fat. The remainder contained crude fiber and a vitamin mix. At 60–80 d of age, female mice were placed individually together with db/+ males and mating was confirmed by the presence of a copulatory plug the next morning, designated d 0 of gestation. In addition to ad libitum-fed db/+ mice, two different levels of caloric restriction were performed in pregnant db/+ mice. In one study, pair feeding was accomplished by measuring the food intake of the ad libitum-fed wild-type +/+ pregnant mice every 24 h and presenting this amount of food to the pair-fed db/+ mice from d 0 of gestation (pair-fed intake was 87% of ad libitum db/+ intake). In a second group, energy intake in db/+ mice was reduced to 70% of ad libitum-fed db/+ mice (70% diet) begun on d 10 of pregnancy. Body weight and food intake of each mouse was recorded daily between 0900 h and 1000 h. For adult studies, the offspring from +/+ mothers and ad libitum-fed db/+ pregnancies were genotyped and the wild-type pups allowed to reach maturity (24 wk) before study. All procedures were approved by the University of Colorado Health Sciences Center Animal Care and Use Committee.
Glucose tolerance test.
Glucose tolerance tests were carried out in conscious, unrestrained mice before pregnancy and on randomly selected mice on d 17 of pregnancy. The mice were fasted for 6 h and injected intraperitoneally with glucose (2 g/kg body weight). Blood was collected from the tail using capillary tubes. Plasma glucose was measured in whole blood by colorimetric glucose oxidase assay (Sigma Chemical, St. Louis, MO, U.S.A.). Insulin was detected using commercial mouse RIA kits (Linco, St. Charles, IL, U.S.A.). Assays were conducted in duplicate and the intra-assay coefficient of variation was <5%.
Cesarean section and fetal tissue collection.
On d 19 of gestation, mice were anesthetized with ketamine (100 mg/kg), acepromazine (10 mg/kg), and xylazine (100 mg/kg). The abdominal cavity was opened and the fetuses removed from the uterus. The pups were weighed and the placenta, liver, and brain were obtained and frozen immediately on dry ice. Livers were weighed and maternal fat mass was determined by the combined weight of dissected fat from the mesenteric, gonadal, retroperitoneal, and dorsal fat pads. The pups were genotyped as carrying the heterozygous mutation (db/+) or wild-type (WT) as described previously (23).
Insulin challenge test in adult animals.
The glucose tolerance test reflects the net result of glucose disposal into peripheral tissues as well as the ability of the pancreatic islets to secrete insulin, and the ability of insulin to inhibit hepatic glucose production. We therefore performed an insulin challenge test as a more direct test of insulin's ability to acutely stimulate glucose disposal on d 18 of gestation as described previously (23). Mice were fasted 6 h and injected intraperitoneally with insulin (0.75 U/kg body weight Humulin R; Eli Lilly, Indianapolis, IN, U.S.A.) and blood was sampled from the tail at 15, 30, and 60 min using capillary tubes. Plasma glucose was measured as above. Later, in a series of experiments in the adult offspring, litters were culled to six per group and the fetuses allowed to reach 24 wk of age. To measure insulin signaling, an acute insulin injection protocol was used to determine maximal insulin stimulation of signaling proteins in liver in vivo, as described previously (15). Mice were anesthetized with ketamine (100 mg/kg), acepromazine (10 mg/kg), and xylazine (100 mg/kg). The abdominal cavity was opened and the portal vein exposed. A 50-μL blood sample was obtained for measurement of glucose and insulin, followed by the injection of a maximal bolus of insulin (10 U/kg body weight in 100 μL saline) into the portal vein. Within 5 min, the liver was excised and frozen immediately. The samples were stored at −80°C until analysis.
Western blot analysis.
Liver homogenates were prepared and protein resolved by 7% SDS-PAGE and transferred to polyvinylidene difluoride membrane using a Mini Trans-Blot Transfer cell (Bio-Rad, Hercules, CA, U.S.A.). The membranes were blocked with 5% nonfat milk in Tris-buffered saline (TBS)-T for 1 h at room temperature and probed overnight at 4°C with anti-IRβ (1:1000, Santa Cruz Biochemicals, Santa Cruz, CA, U.S.A.), anti-IRS-1 (1:1000, Transduction Laboratories, Lexington, KY, U.S.A.), anti-p85α (1:2000, Upstate Biotechnology, Lake Placid, NY, U.S.A.), anti-IRS-2 (1:20, 000, Upstate Biotechnology), anti-GLUT1, GLUT2 (1:4000, Chemicon International, Temecula, CA, U.S.A.), Phospho-Akt (Ser473) or Phosph-p70S6k (Thr421/Ser424) antibody (1:1000, Cell Signaling Technology, Beverly, MA, U.S.A.), or antiphosphotyrosine antibody (1:2000, Upstate Biotechnology). The membranes were washed in TBS-T and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:20,000 dilution in TBS-T, Bio-Rad) for 1 h at room temperature. The bands were visualized with enhanced chemiluminescence (ECL, Amersham Pharmacia Biotech, Arlington Heights, IL, U.S.A.) and exposed to Kodak BIOMAX films (Eastman Kodak, Rochester, NY, U.S.A.). The bands were quantified using a SciScan 500 (USB, Cleveland, OH, U.S.A.) with 50 μg of a normal mouse skeletal muscle protein sample to control for gel-to-gel variation. Each sample was analyzed an average of three separate times involving different gels.
Dual-energy x-ray.
At age 24 wk, mice were subjected to dual-energy x-ray absorptiometry (DEXA) to measure body composition using a densitometer specifically designed for small animals (PIXImus2, Lunar Corp., Madison, WI, U.S.A.), as published previously (24). Before measurements, calibration of the instrument was conducted using a quality control phantom provided by the manufacturer. Mice were weighed after a 4-h fast, anesthetized with Avertin (2,2,2-tribromoethanol, 400 mg/kg), and scanned. Mice were placed on the imaging tray in a prostrate fashion with the front and back legs extended away from the body. Because of the small imaging area (80 × 65 mm), the total length of the animal often exceeded the length of the image. Therefore, the body was maintained in the image area, and much of the head was placed outside the image area. The head and upper portion of the shoulders were excluded from all analyses of body composition. All DEXA determinations and analyses were performed by one investigator (L.Q.). The coefficients of variation from a previous PIXImus model for bone mass density, fat mass, and bone-free lean mass have been reported as 0.84%, 2.20%, and 0.86%, respectively (24). The coefficients of variation for the PIXImus model used in this study were similar (data not shown).
G-6-Pase.
G-6-Pase was assayed in intact microsomes prepared from adult livers. Pieces of freeze-clamped liver were taken randomly, reduced to powder under liquid nitrogen, and rapidly homogenized by sonication in 10 mM N-2-hydroxyethylpiperazine-N ′-2-ethansulfonic acid (HEPES) and 0.25 M sucrose, pH 7.4. Microsomes were extracted by the procedures of Daniele et al.(25). The resulting supernatant was then centrifuged for 30 min at 100,000 ×g at 4°C. The pelleted microsomes were re-suspended in 10 mM HEPES, 0.25 M sucrose. G-6-Pase was assessed on intact microsomes at glucose-6-phosphate concentrations of 1, 2.5, and 10 mM. Total protein was determined by the Bradford method. Integrity of the microsomal membranes was assessed using mannose-6-phosphate (1 mM) as substrate in the G-6-Pase assay, because this substrate is neither bound nor transported by the glucose-6-phosphate transporter but has equivalent reactivity with the catalytic subunit (26). Integrity was >90% in all preparations. Nonspecific phosphatase activity was determined using paranitrophenylphosphate and this value was subtracted from that determined with glucose-6-phosphate.
Statistical analysis.
Results are presented as means ± SEM for the indicated number of mice. Comparisons between groups were made using one-way ANOVA and unpaired t test. Statistical significance was set at p < 0.05.
RESULTS
Changes in energy intake during pregnancy.
Energy intake was not different before pregnancy in WT and db/+ mice. However, during pregnancy, db/+ mice ate significantly more (p < 0.05) by 11%, 14%, and 18% during early (d 0–5), mid- (d 6–11), and late (d 12–16) gestation, respectively (Table 1).
Changes in maternal weight gain and birth weight.
Maternal weight gain was 33% greater (p < 0.05) in ad libitum-fed db/+ mice compared with ad libitum-fed WT controls (Table 2). Pair-feeding db/+ mice to WT controls reduced energy intake by 18% (p < 0.05) and normalized maternal weight gain. Reducing intake further in a second group of db/+ mice to 70% of ad libitum-fed db/+ mice decreased maternal weight gain by 63% (p < 0.05) compared with db/+ ad libitum fed mice, and below WT controls by 44% (p < 0.05). Fetuses were delivered by cesarean section on d 18. The number of fetuses in db/ +, WT, pair-fed, and 70% dams were not different. We genotyped the fetuses for the db mutation by PCR-restriction fragment length polymorphism and there were no significant differences between +/+ and db/+ fetuses from the same litter in terms of birth weight, liver size, or placenta size (data not shown). We therefore grouped the data from each litter together for analysis. Consistent with GDM, the fetuses from ad libitum-fed db/+ mothers were significantly heavier by 6.5% (p < 0.05) compared with fetuses from WT mothers, regardless of fetal genotype. Pair-feeding db/+ mothers reduced fetal weight (p < 0.05) to levels similar to WT mice. Caloric restriction to 70% in the db/+ mothers reduced fetal weight by 10.4% below that of offspring from db/+ ad libitum fed mice (p < 0.05), and by 4.6% below that of offspring from WT mice (p < 0.05).
Effects of caloric restriction on maternal glucose tolerance.
Figure 1 shows that in the nonpregnant state glucose levels were normal in db/+ mice during a glucose tolerance test, however, insulin was 91% and 163% higher at 30 and 60 min, respectively, compared with WT mice (p < 0.05). As shown in Figure 2, pregnancy triggered profound glucose intolerance in ad libitum-fed db/+ mice. Glucose levels were significantly elevated throughout the glucose tolerance test compared with pregnant WT controls (p < 0.01). Fasting and post-challenge insulin levels were also increased by 30–84% (p < 0.05) in db/+ compared with WT mice. Pair-feeding db/+ mice reduced glucose levels by 17% and by 33% (p < 0.05) at 30 min and 60 min compared with ad libitum-fed db/+ mice. Pair-feeding also reduced fasting and postprandial insulin levels by 56–71% (p < 0.05) compared with db/+ ad libitum-fed, and by 61% (p < 0.05) compared with WT mice. Caloric restriction to 70% reduced fasting glucose by 33% (p < 0.05) compared with pair-fed mice, and by 43% (p < 0.05) compared with ad libitum-fed db/+ mice. Caloric restriction to 70% decreased glucose levels by 29% at 30 min (p < 0.05) compared with db/+ pair fed mice. The insulin levels were lower at all time points (p < 0.05) compared with WT mice, or db/+ pair-fed animals at 30 min (p < 0.05).

Serum glucose and insulin concentration during an intraperitoneal glucose tolerance test in WT, db/+ mice in the nonpregnant state. Mice were fasted 6 h and given a 2 g/kg body weight glucose load at time 0 and glucose and insulin levels were determined before and 30 and 60 min after injection. Values are means ± SE for nine WT mice and 20 db/+ mice. *p < 0.05.

Serum glucose and insulin concentration during an intraperitoneal glucose tolerance test in pregnant WT, db/+, db/+ pair-fed, and db/+ mice fed 70% diet. Selected mice were fasted 6 h and injected with 2g/kg body weight glucose load at time 0 and insulin levels were determined before and 30 and 60 min after injection. Values are means ± SEM for six to 10 mice per group. *p < 0.05, WT vs db/+; §p < 0.05 WT vs pair-fed; ¶p < 0.05 WT vs 70% diet; †p < 0.05 db/+ vs pair-fed; ‡p < 0.05 db/+ vs 70% diet; **p < 0.05 pair-fed vs 70% diet. Values are means ± SE.
Insulin challenge test.
As a more direct test of insulin's ability to acutely stimulate glucose disposal, we carried out an acute insulin challenge test in the mice on day 18 of pregnancy (Fig. 3). The db/+ mice showed 48–75% higher (p < 0.05) glucose levels, indicative of insulin resistance, compared with WT mice. Pair feeding in db/+ mice lowered glucose levels compared with db/+ ad libitum-fed mice, but this was not statistically significant. The 70% diet decreased glucose levels by 47–69% below ad libitum-fed db/+ mice (p < 0.05) and by 62% compared with pair-fed mice at the 60-min time point (p < 0.05), without affecting fasting glucose.

Insulin challenge test in WT, db/+, db/+ mice pair-fed, and db/+ mice fed 70% diet . Selected mice were fasted 6 h on d 18 of pregnancy and injected with 0.75 U/kg body weight regular insulin at time 0 and glucose level determined before and 15, 30, and 60 min after injection. Results are expressed as percentage of blood glucose concentration before insulin injection. Values are means ± SE for six to 10 mice per group. *p < 0.05, between WT and db/+ mice; ‡p < 0.05, between db/+ and 70% diet mice; **p < 0.05, between pair-fed and 70%-fed mice.
Effect of GDM and caloric restriction on insulin-signaling proteins in fetal livers.
There was no significant effect of GDM or caloric restriction on GLUT1, GLUT2, IRS-2, and p85α PI 3-kinase protein levels (not shown) in fetal livers. However, the level of insulin receptor protein was significantly lower by 31% (p < 0.05) in offspring from ad libitum-fed db/+ mothers compared with offspring from WT mothers (Fig. 4). Pair-feeding db/+ mice restored fetal insulin receptor protein levels to WT controls. Caloric restriction in db/+ mothers to 70% reduced the level of insulin receptor protein in liver below the level of WT offspring by 28% (p < 0.05).

Representative immunoblot and quantification of insulin receptor protein in fetal livers. Fetuses were removed by cesarean section on d 18 of pregnancy from WT, db/+ ad libitum-fed, db/+ pair-fed, and db/+ 70%-fed mothers. Fetuses were killed and livers were removed and frozen at −80°C until analysis. DNA was extracted and fetuses genotyped according to procedures outlined under “Methods.” The livers from WT pups were homogenized and equal amounts of protein separated by 7% SDS-PAGE and analyzed by Western blotting with anti-GLUT1, -GLUT2, -IRβ, -IRS-2, anti-PI 3-kinase (85α kD subunit). The values shown are the mean ± SE (n = 10–12 per group) for IR-β subunit based on scanning densitometry (arbitrary units), compared with an internal standard to control for gel-to-gel variation.
Changes in body weight, percentage fat, insulin, and glucose in WT and GDM offspring at 6 mo of age.
To determine the effect of GDM on insulin resistance in later life, the offspring from WT and ad-libitum fed db/+ mothers were genotyped and groups of males and female wild-type were studied at 24 wk of age (Table 3). Total body weight was similar in offspring from db/+ and WT mothers, however, percentage body fat was significantly greater (p < 0.05) only in the female offspring from GDM mothers as determined by total-body DEXA analysis. Fasting insulin levels in GDM offspring were also 2-fold greater in females only (p < 0.05). The IUGR fetuses were not studied as adults.
G-6-Pase activity.
To determine whether GDM affected the enzymatic capacity for hepatic glucose production, we examined the activity of G-6-Pase, the terminal enzyme of gluconeogenesis and glycogenolysis. G-6-Pase activity was significantly increased in the GDM offspring by 6-fold (p < 0.01) at 0.5 mM glucose-6-phosphate concentration and by 25% (p < 0.05) at 10 mM G-6-Pase concentration (Fig. 5). Nonspecific phosphatase activity as determined by using paranitrophenylphosphate as a substrate was not different (data not shown).

G-6-Pase activity in livers from WT and GDM offspring at 6 mo of age. Livers were removed and frozen at −80°C until analysis. Intact microsomes were prepared and G-6-Pase activity assayed as described in “Methods.” Values are means ± SE (n = 4–5 animals per group). *Significantly different from WT offspring, p < 0.05.
Effects of GDM on insulin signaling in adult livers.
The increased G-6-Pase activity in female offspring from GDM mothers, despite hyperinsulinemia, led us to investigate whether aspects of the insulin receptor signal cascade were altered in response to insulin in livers of GDM offspring. We measured the expression and tyrosine phosphorylation of the insulin receptor, IRS-2, p85α PI3-kinase subunit, and the activation of downstream serine/threonine kinases Akt-1 and p70S6 kinase in response to insulin (Fig. 6). There were no differences in total expression or tyrosine phosphorylation of the insulin receptor, IRS-2, and p85α subunit of PI-3 kinase in GDM offspring compared with WT controls (data not shown). However, the levels of insulin-stimulated 473Ser-Akt phosphorylation were 65% lower (p < 0.05) in offspring from GDM mice. The total amount of Akt was not different in the livers, suggesting this did not contribute to lower levels of phosphorylated Akt. To determine whether signals immediately downstream from Akt were reduced, Western blotting of p70 S6 kinase was performed. There was no significant difference in the amount of p70 S6 kinase, however, phosphorylation on 424Ser-p70 S6 kinase in response to insulin was decreased by 55% (p < 0.05).

Representative immunoblots and quantification of insulin signaling in intact livers from WT and GDM offspring at 6 mo of age. Female mice were fasted for 6 h, anesthetized, and insulin injected into the portal vein (1 mU/kg body weight) as outlined in “Methods.” Livers were removed within 2 min after insulin treatment. Proteins were analyzed for tyrosine phosphorylation by immunoprecipitation with an antiphosphotyrosine antibody followed by immunoblotting for IR, IRS-2, and p85α subunit of PI 3-kinase. Akt and p70S6 Kinase protein levels and serine phosphorylation were determined in protein extracts using phospho-specific antibodies as described under “Methods.” The results are expressed as arbitrary units relative to WT mice. Data are means ± SE. *p < 0.05 between WT and GDM offspring, n = 4–6 samples per group. Open bars are results from WT mothers, solid bars depict the response in GDM offspring.
DISCUSSION
GDM is associated with extreme insulin resistance, increased adiposity, and a greater risk for producing obesity and diabetes in the offspring. Here we demonstrate that excess caloric intake during pregnancy triggers spontaneous GDM in C57BL/KsJ-Leprdb/+mice. Our results show that pair-feeding the Leprdb/+ mothers to the energy intake in WT +/+ mice significantly reduces maternal weight gain and prevents fetal overgrowth in this model. More stringent caloric restriction was associated with improved insulin sensitivity below WT controls, but resulted in fetal IUGR. These results imply that limiting maternal energy intake may be necessary for reducing maternal insulin resistance, but may pose an increased risk for fetal undergrowth. It is well known that limitations in substrate availability (e.g. mothers with hypoglycemia), during pregnancy are more likely to result in small for gestational age neonates (27, 28).
Interestingly, we found significant decreases in insulin receptor protein levels in livers from both IUGR and in fetuses from ad libitum-fed GDM mothers. These results are similar to Mulay et al.(29), who reported reduced 125I-insulin binding to partially purified liver membranes from both IUGR and streptozotocin-induced diabetic fetuses. This is in contrast to observations made in other fetal tissues such as rabbit heart (30), lung (31), or macrophages (32), where the insulin receptor concentration is either up-regulated or unchanged in response to high insulin. Earlier studies reported significant decreases in circulating insulin and glucagon in caloric-restricted rat fetuses, however, maternal food restriction did not affect glucagon or insulin binding to liver membranes in the fetus (33, 34). The mechanisms underlying the similar decreases in expression of fetal insulin receptors in liver from GDM and IUGR fetuses are unknown.
To determine whether adult offspring of GDM mothers were insulin resistant, we focused on identifying postnatal changes in the liver of the adult animals from GDM mothers. At 24 wk of age, only females from ad libitum-fed GDM mothers were hyperinsulinemic and had significantly increased body fat compared with females born from WT mothers. Van Assche et al.(35, 36) found that only adult offspring of moderately diabetic mothers (using streptozotocin) had a deficient β-cell response to glucose stimulation, whereas adults from severely diabetic mothers were insulin resistant. When females from these two groups became pregnant, they developed GDM and their fetuses display the same biochemical phenotypes found in the first generation. This transmission occurred only in females of diabetic mothers suggesting that metabolic (androgen or estrogen) differences between males and females may contribute to hyperinsulinemia and increased body fat as a result of maternal diabetes in these models.
Another important finding of the present study is that the female offspring from GDM mothers had elevated hepatic G-6-Pase activity at each level of G-6-Pase level tested, despite chronic hyperinsulinemia. We did not detect fasting hyperglycemia in the adult GDM offspring, however, reduced insulin signaling and increased G-6-Pase have their greatest effects in the insulin-stimulated state (e.g. feeding), which would be manifest by reduced suppression of hepatic glucose production. In the basal state, these impairments will not result in overproduction of glucose or fasting hyperglycemia because of compensatory fasting hyperinsulinemia. In support of this concept is the finding that GDM offspring had a specific defect in insulin signaling at the level of serine/threonine kinase Akt/PKB phosphorylation. Because activation of Akt is necessary for insulin-induced suppression of hepatic glucose output, a decrease in Akt phosphorylation, increased G-6-pase activity, and hyperinsulinemia suggest that the livers were very likely insulin resistant. Most studies suggest that insulin signaling via Akt is a critical intermediate in insulin's ability to suppress activity of the PEPCK and G-6-Pase promoters (37–39). In mice with a gene deletion in Akt2, there is reduced insulin suppression of hepatic glucose production accompanied by hyperinsulinemia with normoglycemia (18). Our data support the idea that the specific decrease in Akt phosphorylation in livers of adult offspring may be a mechanism for reduced insulin-dependent physiologic events, such as suppression of hepatic glucose production, a defect associated with susceptibility to type II DM.
CONCLUSION
In summary, our results suggest that there is a strong effect of maternal GDM on fetal growth and this effect reveals itself in the subsequent development of obesity and insulin resistance in the adult liver in female offspring. The insulin resistance is associated with a specific abnormality in liver Akt phosphorylation and an increase in hepatic G-6-Pase activity. The defect in Akt activation by insulin was also associated with greater percentage fat and fasting hyperinsulinemia, both hallmarks of insulin resistance. The expression and phosphorylation of the insulin receptor was unaffected in the liver of mature GDM offspring. These data suggest that the decrease in insulin receptors in the fetal liver may not participate in the defect of insulin signaling in the adult, but could play a significant role in insulin resistance during other key periods of development. Such influences could alter the normal balance of expression levels of genes that regulate substrate metabolism and energy deposition leading to increased body fat and hepatic insulin resistance in the adult offspring.
Abbreviations
- GDM:
-
gestational diabetes mellitus
- IGT:
-
impaired glucose tolerance
- Lepr db/+ :
-
leptin receptor deficient
- WT:
-
wild-type
- IUGR:
-
intrauterine growth retardation
- IR-α:
-
insulin receptor α
- IRS-1:
-
insulin receptor substrate-1
- GLUT:
-
glucose transporter
- PI 3-kinase:
-
phosphotidylinositol-3-kinase
- Akt (c-PkB):
-
related to A kinase, protein kinase B
- G-6-Pase:
-
glucose-6-phosphatase
- HGP:
-
hepatic glucose production
References
Barker DJ 1995 Intrauterine programming of adult disease. Mol Med Today 1: 418–423
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD 1991 Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303: 1019–1022
Pettitt DJ, Nelson RG, Saad MF, Bennett PH, Knowler WC 1993 Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care 16: 310–314
Dandrow RV, O'Sullivan JB 1966 Obstetric hazards of gestational diabetes. Am J Obstet Gynecol 96: 1144–1147
Stevenson DK, Hopper AO, Cohen RS, Bucalo LR, Kerner JA, Sunshine P 1982 Macrosomia: causes and consequences. J Pediatr 100: 515–520
Rizzo T, Metzger BE, Burns WJ, Burns K 1991 Correlations between antepartum maternal metabolism and child intelligence. N Engl J Med 325: 911–916
Onyeije CI, Divon MY 2001 The impact of maternal ketonuria on fetal test results in the setting of postterm pregnancy. Am J Obstet Gynecol 184: 713–718
Gewolb IH, Barrett C, Wilson CM, Warshaw JB 1982 Delay in pulmonary glycogen degradation in fetuses of streptozotocin diabetic rats. Pediatr Res 16: 869–873
Ishizuka T, Klepcyk P, Liu S, Panko L, Gibbs EM, Friedman JE 1999 Effects of overexpression of human GLUT4 gene on maternal diabetes and fetal growth in spontaneous gestational diabetic C57BLKS/J Lepr(db/+) mice. Diabetes 48: 1061–1069
Kaufmann RC, Amankwah KS, Dunaway G, Maroun L, Arbuthnot J, Roddick JW 1981 An animal model of gestational diabetes. Am J Obstet Gynecol 141: 479–482
Lawrence S, Warshaw J, Nielsen HC 1989 Delayed lung maturation in the macrosomic offspring of genetically determined diabetic (db/+) mice. Pediatr Res 25: 173–179
Kahn CR 1994 Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 43: 1066–1084
Shepherd PR, Withers DJ, Siddle K 1998 Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 333: 471–490
Czech MP, Corvera S 1999 Signaling mechanisms that regulate glucose transport. J Biol Chem 274: 1865–1868
Sun Y, Liu S, Ferguson S, Wang L, Klepcyk P, Yun JS, Friedman JE 2002 Phosphoenolpyruvate carboxykinase over-expression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J Biol Chem 8: 6
Bevan P 2001 Insulin signalling. J Cell Sci 114: 1429–1430
Hall RK, Granner DK 1999 Insulin regulates expression of metabolic genes through divergent signaling pathways. J Basic Clin Physiol Pharmacol 10: 119–133
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ 2001 Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292: 1728–1731
Burgering BM, Coffer PJ 1995 Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376: 599–602
Nakae J, Kitamura T, Silver DL, Accili D 2001 The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 108: 1359–1367
Forsen T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D 2000 The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med 133: 176–182
Rich-Edwards JW, Colditz GA, Stampfer MJ, Willett WC, Gillman MW, Hennekens CH, Speizer FE, Manson JE 1999 Birthweight and the risk for type 2 diabetes mellitus in adult women. Ann Intern Med 130: 278–284
Yamashita H, Shao J, Ishizuka T, Klepcyk PJ, Muhlenkamp P, Qiao L, Hoggard N, Friedman JE 2001 Leptin administration prevents spontaneous gestational diabetes in heterozygous Leprdb/+: effects on placental leptin and fetal growth. Endocrinology 142: 2888–2897
Barbour LA, Shao J, Qiao L, Pulawa LK, Jensen DR, Bartke A, Garrity M, Draznin B, Friedman JE 2002 Human placental growth hormone causes severe insulin resistance in transgenic mice. Am J Obstet Gynecol 186: 512–517
Daniele N, Rajas F, Payrastre B, Mauco G, Zitoun C, Mithieux G 1999 Phosphatidylinositol 3-kinase translocates onto liver endoplasmic reticulum and may account for the inhibition of glucose-6-phosphatase during refeeding. J Biol Chem 274: 3597–3601
Minassian C, Mithieux G 1994 Differential time course of liver and kidney glucose-6 phosphatase activity during fasting in rats. Comp Biochem Physiol B Biochem Mol Biol 109: 99–104
Piper JM, Field NT, Higby K, Elliott BD, Langer O 1996 Maternal-fetal glucose metabolism and fetal growth retardation. Is there an association?. J Reprod Med 41: 761–766
Ogata ES, Paul RI, Finley SL 1987 Limited maternal fuel availability due to hyperinsulinemia retards fetal growth and development in the rat. Pediatr Res 22: 432–437
Mulay S, Philip A, Solomon S 1983 Influence of maternal diabetes on fetal rat development: alteration of insulin receptors in fetal liver and lung. J Endocrinol 98: 401–410
Devaskar SU, Szewczyk K, Devaskar UP 1987 The fetal heart insulin receptor responds differently to varying plasma insulin concentrations. Dev Pharmacol Ther 10: 153–162
Neufeld ND, Scott M, Kaplan SA 1980 Ontogeny of the mammalian insulin receptor. Studies of human and rat fetal liver plasma membranes. Dev Biol 78: 151–160
Neufeld ND, Kaplan SA, Lippe BM 1981 Monocyte insulin receptors in infants of strictly controlled diabetic mothers. J Clin Endocrinol Metab 52: 473–476
Alvarez E, Fernandez S, Blazquez E 1986 Effect of maternal food restriction on circulating insulin and glucagon levels and on liver insulin and glucagon binding sites of fetal and suckling rats. Diabetes Metab 12: 337–345
Alvarez E, Blazquez E 1987 Lack of insulin effect on its own receptors in fetal rat hepatocytes. Horm Metab Res 19: 458–463
Aerts L, Holemans K, Van Assche FA 1990 Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev 6: 147–167
Holemans K, Aerts L, Van Assche FA 1991 Evidence for an insulin resistance in the adult offspring of pregnant streptozotocin-diabetic rats. Diabetologia 34: 81–85
Sutherland C, O'Brien RM, Granner DK 1995 Phosphatidylinositol 3-kinase, but not p70/p85 ribosomal S6 protein kinase, is required for the regulation of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by insulin. Dissociation of signaling pathways for insulin and phorbol ester regulation of PEPCK gene expression. J Biol Chem 270: 15501–15506
Gabbay RA, Sutherland C, Gnudi L, Kahn BB, O'Brien RM, Granner DK, Flier JS 1996 Insulin regulation of phosphoenolpyruvate carboxykinase gene expression does not require activation of the Ras/mitogen-activated protein kinase signaling pathway. J Biol Chem 271: 1890–1897
Barthel A, Schmoll D, Kruger KD, Bahrenberg G, Walther R, Roth RA, Joost HG 2001 Differential regulation of endogenous glucose-6-phosphatase and phosphoenolpyruvate carboxykinase gene expression by the forkhead transcription factor FKHR in H4IIE-hepatoma cells. Biochem Biophys Res Commun 285: 897–902
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by National Institute of Child Health and Human Development Grant P50 11089-20 and National Institutes of Health Grant DK55386.
Rights and permissions
About this article
Cite this article
Yamashita, H., Shao, J., Qiao, L. et al. Effect of Spontaneous Gestational Diabetes on Fetal and Postnatal Hepatic Insulin Resistance in Leprdb/+ Mice. Pediatr Res 53, 411–418 (2003). https://doi.org/10.1203/01.PDR.0000049667.58071.7D
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000049667.58071.7D
This article is cited by
-
A fresh look to the phenotype in mono-allelic likely pathogenic variants of the leptin and the leptin receptor gene
Molecular and Cellular Pediatrics (2021)
-
Vitamin B6 deficiency disrupts serotonin signaling in pancreatic islets and induces gestational diabetes in mice
Communications Biology (2021)
-
Poorly controlled diabetes during pregnancy and lactation activates the Foxo1 pathway and causes glucose intolerance in adult offspring
Scientific Reports (2019)
-
Late Cognitive Consequences of Gestational Diabetes to the Offspring, in a New Mouse Model
Molecular Neurobiology (2019)
-
Absence of a gestational diabetes phenotype in the LepRdb/+ mouse is independent of control strain, diet, misty allele, or parity
Scientific Reports (2017)
