
Abstract
Ocular nonnephropathic cystinosis, a variant of the classic nephropathic type of cystinosis, is an autosomal recessive lysosomal storage disorder characterized by photophobia due to corneal cystine crystals but absence of renal disease. We determined the molecular basis for ocular cystinosis in four individuals. All had mutations in the cystinosis gene CTNS, indicating that ocular cystinosis is allelic with classic nephropathic cystinosis. The ocular cystinosis patients each had one severe mutation and one mild mutation, the latter consisting of either a 928 G→A (G197R) mutation or an IVS10–3 C→G splicing mutation resulting in the insertion of 182 bp of IVS10 into the CTNS mRNA. The mild mutations appear to allow for residual CTNS mRNA production, significant amounts of lysosomal cystine transport, and lower levels of cellular cystine compared with those in nephropathic cystinosis. The lack of kidney involvement in ocular cystinosis may be explained by two different mechanisms. On the one hand (e.g. the G197R mutation), significant residual cystinosin activity may be present in every tissue. On the other hand (e.g. the IVS10–3 C→G mutation), substantial cystinosin activity may exist in the kidney because of that tissue's specific expression of factors that promote splicing of a normal CTNS transcript. Each of these mechanisms could result in minimally reduced lysosomal cystine transport in the kidneys.
Similar content being viewed by others

Main
Cystinosis is an autosomal recessive lysosomal storage disorder in which the disulfide amino acid cystine accumulates to crystal-forming levels within cellular lysosomes (1). Patients with the classic infantile nephropathic form of this disease are normal at birth but typically present with renal tubular Fanconi syndrome in infancy, characterized by failure to thrive, dehydration, polyuria and polydipsia, acidosis, hypophosphatemic rickets, and hypokalemia (1, 2). Hypothyroidism and photophobia due to corneal cystine crystals develop at variable times, but renal glomeruli lose function progressively, resulting in kidney failure at approximately 10 y of age. After a renal allograft procedure, cystine accumulation continues to destroy the nonrenal organs of cystinosis patients (3), frequently causing a distal vacuolar myopathy (4, 5), swallowing difficulty (6), pancreatic endocrine (7) and exocrine (8) insufficiency, CNS deterioration (9, 10), testicular dysfunction (11), and ophthalmic complications that include severe retinal dysfunction (12). The treatment of cystinosis consists of replacement of renal losses and cystine depletion by using cysteamine orally (13–17) or topically (18, 19).
Variants of classic nephropathic cystinosis contribute to a broad spectrum of disease severity. Individuals with intermediate, or juvenile, cystinosis have the same signs and symptoms of classic cystinosis only at a later age (1). Patients with ocular nonnephropathic cystinosis, first reported in 1957 (20) and termed “benign” or “adult” cystinosis, never suffer renal disease and do not exhibit a retinal pigment abnormality but do have crystals in their cornea and bone marrow (1). Of the approximately 15 ocular cystinosis patients reported, most presented in childhood with mild photophobia or with corneal crystals on routine ophthalmologic examination. No ocular cystinosis patient has experienced any consequence of the disorder besides photophobia. Ocular cystinosis patients generally have leukocyte cystine levels of 1–3 nmol half-cystine per milligram of protein, compared with 3–23 for classical patients (normal, ≤0.2). Based upon fibroblast complementation studies, ocular cystinosis has been considered allelic with the classic nephropathic disease (21).
The basic defect in cystinosis involves deficiency of a cystine transporter in the lysosomal membrane (22–24). The normal cystine carrier displays characteristics of both egress (22, 23) and countertransport (25), a phenomenon in which a small radiolabeled ligand appears to traverse a membrane against its concentration gradient. The lysosomal cystine carrier is presumably encoded by the cystinosis gene CTNS, mapped to chromosome 17p in 1995 (26) and isolated in 1998 (27). CTNS is known to contain 12 exons distributed across ∼23 kb of genomic DNA; the CTNS gene product cystinosin has 367 amino acids with seven predicted transmembrane domains and eight potential glycosylation sites (27). To date, 31 distinct mutations in CTNS have been reported (27, 28), but the most common is a 65-kb deletion found frequently in patients of northern European descent. This deletion, now known to be 57 kb (J. Touchman and E. Green, personal communication), was found in 56% of the alleles of 108 cystinosis patients seen in the United States (29).
We performed mutation analysis of the CTNS gene on four ocular cystinosis patients. The results indicate that the nonnephropathic variant is, indeed, allelic with the classic disorder. Moreover, ocular cystinosis patients exhibit a unique array of novel mutations combined with known classic mutations.
METHODS
Patients.
All patients and their families were enrolled in a protocol approved by the Institutional Review Board at the National Institutes of Health Clinical Center or the University of California, San Diego Center for the Health Sciences. The patients were examined and treated for various durations at these institutions.
Clinical biochemical studies.
Leukocyte cystine values were determined using the cystine-binding protein method (30). The severity of the renal tubular reabsorption defect was gauged by the Fanconi syndrome index; a measure of the daily urinary excretion of 21 amino acids per kilogram body weight (31) was used (28). Patients were also evaluated using a recently devised clinical severity scale involving five parameters, i.e. age at presentation, leukocyte cystine value, Fanconi syndrome index, age at renal failure, and age of nonrenal complications. According to this scale, a value of 1 is extremely mild and a value of 3 is very severe. Typical nephropathic cystinosis patients have a value of 2.0.
Molecular studies.
Genomic DNA of each patient was initially screened for mutations by performing SSCP analysis on exons 3–12 of CTNS as previously described (28). Any exons showing abnormal bands were subjected to direct DNA sequencing using the ABI Prism dRhodamine Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) according to the manufacturer's recommendations.
Detection of the 57-kb common deletion in the heterozygous state was accomplished using primers flanking the deletion. This gave a 423-bp product if the deletion was present, and no product if the deletion was absent (29). In this multiplex PCR reaction, primers for the polymorphic marker D17S829 were included to verify the presence of a nondeleted allele.
Analysis of the polymorphic microsatellite marker D17S829 in cases 3 and 4 was performed as previously described (28). Northern blot analysis was performed as previously described (28) using human CTNS cDNA (exons 3–10) random primer labeled with α[32P]-dCTP (DuPont/NEN, Wilmington, DE) as probe.
RT-PCR was performed after total RNA extraction from cultured fibroblasts by using Trizol reagent (Life Technologies, Grand Island, NY). cDNA was synthesized by reverse transcription by using 5 μg of total RNA according to the manufacturer's protocol (GIBCO-BRL, Grand Island, NY). PCR amplification was performed using 2 μL of the first-strand cDNA, 1x PCR buffer (GIBCO-BRL), 1.5 mM MgCl2, 200 μM dNTP, 0.2 μM of each primer, and 2.5 U Taq DNA polymerase in a final volume of 50 μL. After an initial denaturation step at 94°C for 4 min, PCR was conducted for 30 cycles, each with a denaturation step at 94°C for 30 s, annealing at 55°C for 40 s, and extension at 72°C for 1.5 min. (The extension time was 2.5 min when the entire coding cDNA was amplified.) An elongation step at 72°C for 10 min finalized the procedure. The PCR products were electrophoresed in 1.4% agarose and were stained with ethidium bromide. The primers used for amplification of the coding cDNA were 5′-CCTCTTCCAGTAACATTGAGG-3′ (in exon 2) and 5′-AGAAAAGAGATGGCGGTGTC-3′ (in the 3′ UTR), yielding a 1431-bp product. The primers used to create the probe for Northern blot analysis were 5′-TGAAGCTCGTAGAGAAATGTG-3′ and 5′-GCTTGATGTAGGAGAAGCAG-3′, yielding a 779-bp band. The primers used to amplify the paternal allele of case 1, excluding the maternal allele, were 5′-CTATCCTTGAGCTCCCCG-3′ and 5′-GGTTGGGTCTCCGAAGATC-3′, yielding a 799-bp product.
CASE HISTORIES
Case 1.
This 26-y-old male was patient 2 of a previous report (32). Briefly, he presented with corneal crystals on routine ophthalmologic examination at 6 y 9 mo of age. At that time, a renal biopsy, fixed in absolute ethanol to preserve cystine crystals, showed no crystals, but a bone marrow biopsy showed typical crystals on light microscopy (Fig. 1A) and under birefringent light (Fig. 1B). Serum creatinine was 0.7 mg/dL, growth and bone ages were normal, and there was no evidence of renal tubular Fanconi syndrome. By age 14 y 6 mo, the serum creatinine was 1.0 mg/dL, height was 166 cm (50%), and weight was 73 kg (90%). Renal tubular function was normal. At this time, the leukocyte cystine level was 2.85 nmol half-cystine/mg protein, and the countertransport of cystine across the membranes of leukocyte granular fractions (i.e. lysosomes) was 9% of normal on one occasion and 29% of normal on another. The patient's father had a leukocyte cystine level of 0.52 nmol half-cystine/mg protein and 61 and 73% of the normal lysosomal cystine countertransport. The mother had a leukocyte cystine level of 0.32 nmol half-cystine/mg protein and 66 and 70% of the normal amount of countertransport.

Biopsies of case 1. A, Light microscopy of bone marrow showing hexagonal crystals against a background of normal cells. B, Bone marrow crystals under cross-polarizing light.
At age 20 y, the patient had dense crystals in all layers of his corneas (Fig. 2A). Between 20 and 26 y of age, the patient's creatinine clearance ranged from 101 to 127 mL/min (normal, 90–125 mL/min), and his Fanconi syndrome index ranged from 51 to 72 μmol·kg−1·d−1 (normal, 94 ± 45). His leukocyte cystine ranged from 8 to 16 nmol half-cystine/mg protein, but he never received oral cysteamine therapy. At age 26 y 6 mo, the patient's height was 184 cm (85%) and his weight was 109 kg (>95%). The serum creatinine was 1.0 mg/dL, and two creatinine clearances were 150 and 167 mL/min. Serum electrolytes and blood counts were normal. The thyroxine level was 8.1 μg/dL (normal, 5.0–10.0), and the TSH was 1.61 μIU/mL (normal, 0.4–4.4). Testosterone was 511 ng/dL (normal, 300–950), with an LH of 5 U/L (normal, 2–12). Total serum cholesterol was 193 mg/dL (normal, 100–200 mg/dL). The leukocyte cystines were 14.8 and 18.6 nmol half-cystine/mg protein, and a fibroblast cystine level was 3.7 nmol half-cystine/mg protein. The clinical severity score was 1.2, indicating extremely mild disease.

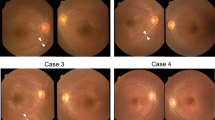
Slit lamp photographs of cornea. A, Case 1 at age 20 showing abundant birefractive corneal crystals. B, Case 3 at age 20 showing moderate crystals. C, Case 4 at age 21 with moderate corneal crystal density.
Case 2.
This 38-y-old woman presented with photophobia at age 38 y but had suffered chronic sensitivity to light. Fluorescent lights, driving into headlights, and peering at flames were especially bothersome. On examination, the patient was 164 cm in height (50%) and weighed 64 kg (75%). Typical corneal and conjunctival crystals were seen by an ophthalmologist, but a nephrologist found no medical evidence of renal tubular or glomerular damage. The serum creatinine was 0.9 mg/dL, with normal electrolytes and blood counts. The urinalysis was normal. The leukocyte cystine level was 1.2 nmol half-cystine/mg protein. The clinical severity score was 1.0, based upon four of the five designated parameters (28).
Case 3.
This 20-y-old woman was found to have corneal crystals on an optometrist's routine examination 6 mo before admission. Crystals observed by an optometrist at age 12 were interpreted as artifact due to cosmetic use. Chronic mild photophobia and a foreign body sensation were increased during her pregnancy 7 mo before admission, but the patient had no other health problems. She never received oral cysteamine therapy. The physical examination was entirely normal except for the presence of moderate crystals in the anterior stroma of the corneas (Fig. 2B). Pigmentation of the retina was normal. The patient was 170 cm tall (85%) and 56.6 kg (50%). Laboratory studies revealed normal serum electrolytes, blood counts, and serum chemistries. The serum creatinine was 0.7 mg/dL, with a clearance of 144 mL/min (normal, 90–125). Two Fanconi syndrome indexes were normal, i.e. 85 and 109 μmol·kg−1·d−1, and leukocyte cystines on 2 days were 1.3 and 1.1 nmol half-cystine/mg protein. The clinical severity score was 1.0, the lowest possible value.
Case 4.
This 21-y-old woman was the first cousin of case 3. She was also diagnosed with corneal crystals on routine ophthalmologic examination and had suffered chronic photophobia. Otherwise, she was an entirely healthy college student. On physical examination, the patient was 164 cm in height (50%) and 77 kg in weight (>90%). Her ophthalmologic examination revealed moderate crystals in the anterior stroma of both corneas (Fig. 2C) and normal pigmentation with a blond fundus. Serum electrolytes, blood counts, and chemistries were normal, with a serum creatinine of 0.7 mg/dL and clearances of 127 and 158 mL/min. Serum thyroxine was 10.3 μg/dL and TSH 1.15 μIU/mL. Two Fanconi syndrome indexes were normal, i.e. 95 and 98 μmol·kg−1·d−1. The leukocyte cystine levels were 2.1 and 2.5 nmol half-cystine/mg protein on consecutive days. The clinical severity score was 1.0.
RESULTS
Mutation analysis.
Mutations were identified on both alleles of case 1. The first mutation was determined after SSCP analysis of exons 3–12 showed an abnormal pattern for exon 5 (Fig. 3A). Direct sequencing of the patient's genomic DNA revealed a heterozygous TCCTT deletion starting at nucleotide 545, causing an I69R amino acid substitution and resulting in a stop codon at position 73. This deletion, previously described in a classic nephropathic cystinosis case (28), was also found in the heterozygous state in the patient's mother but not in his father. The patient's maternal allele, analyzed using cDNA, illustrates the deletion (Fig. 3B).

A, SSCP analysis of exon 5 of CTNS. Lanes 1 and 5, control DNA. Lanes 2 and 4, DNA from nephropathic cystinosis patients showing the normal pattern. Lane 3, DNA from case 1 showing heterozygosity for the normal fragments plus an abnormally migrating pair of bands. B, Sequence of the cDNA of the maternal allele of case 1 (right) compared with normal (left) in the region of exon 5. Note deletion of TCCTT starting at nucleotide 545, after which the sequence is ambiguous due to overlap of the two different contributing alleles.
To search for the patient's second mutated allele, exons 3–12 were completely sequenced from genomic DNA using standard PCR primers (27). This revealed a heterozygous C→G substitution at the −3 position of the acceptor splice site for IVS10 (Fig. 4A, top). The father was a carrier for this mutation. Because the mutation could cause abnormal splicing (33–36), the cDNA in the region was examined. First, a region from exon 5 to 12 was amplified using a 5′ primer that included the TCCTT deletion on the opposite allele; this eliminated amplification of the maternal allele. The products formed included fragments of the expected size (799 bp) as well as a band of 981 bp and an intermediate-sized fragment (Fig. 4B). Analysis of the small band showed the expected sequence. The large fragment contained a 182-bp insertion whose sequence was identical with the last portion of IVS10 (Fig. 4A, bottom). The middle band contained a combination of the two sequences. At the 5′ boundary of the 182-bp insertion within IVS10 was a splice site sequence that had exactly the same degree of resemblance to the consensus sequence, (Py)11NCAG-G, as the normal splice acceptor site. That is, both the cryptic splice site and the normal acceptor site had seven pyrimidines in the 11 nucleotide sequence 5′ to CCAG-G. The cryptic splicing predicts a cystinosin protein with 28 incorrect amino acids inserted, terminating at residue 313. There is inadequate information concerning the structure of cystinosin to infer the impact of this mutation upon the protein's functional domains. A diagram of this abnormal splicing situation is shown in Figure 4C.

Paternal mutation in case 1. A, Sequencing of the genomic DNA of case 1 revealed both the normal C and a mutant G in the −3 position of IVS10 (top). Sequencing of the upper band of lane 3 of (B) revealed 182 bp of IVS10 incorporated into the cDNA between exons 10 and 11 (bottom). B, Agarose gel electrophoresis of cDNA fragments, PCR amplified using a 5′ primer including nucleotides 545–549 to exclude the maternal allele. Lane 1, DNA size markers. Lane 2, negative control containing no DNA. Lane 3, case 1 cDNA template showing three amplified bands. Lane 4, control cDNA template yielding a fragment of the expected size, 799 bp. C, Schematic representation of the paternal mutation of case 1. Normal splicing occurs using the consensus acceptor site. Substitution of a G for a C renders this site less attractive, and a cryptic site 182-bp 5′ of exon 11 serves as the splice acceptor. The abnormal splicing creates the band in (A), which is 182 bp larger than normal.
Two mutations were also identified in case 2. Because SSCP showed no obvious changes, we sequenced the entire coding region of CTNS using both genomic DNA and cDNA. This revealed a G→A substitution at nucleotide 928 (Fig. 5), causing a Gly to Arg change at amino acid 197. Sequencing revealed only the A at this location, indicating that the patient could be homozygous for this mutation. Alternatively, the patient could be hemizygous for the 928 G→A mutation and carry the common 57-kb deletion (27) on the other allele. We studied this possibility by performing PCR amplification using recently identified primers that flank the 57-kb deletion and yield a product only if the deletion is present (29). In case 2, the product was identified (data not shown), demonstrating that the patient carried both the G197R mutation and the 57-kb deletion. No 928 G→A changes were observed in the sequences of 55 other alleles, including 20 normal alleles and 35 alleles containing a different CTNS mutation.

Sequence of a portion of exon 9 of CTNS using as template either normal control DNA (left) or genomic DNA of patient 2 (right). The G→A substitution at nucleotide 928 results in a G197R amino acid change.
Cases 3 and 4 were part of a family whose pedigree is shown in Figure 6. DNA was available from six of the seven members depicted. The two patients with ocular cystinosis, II-1 and II-2, were cases 3 and 4, respectively. For both patients, sequencing of exons 3–12 of genomic DNA revealed the identical 928 G→A substitution found in case 2. Members of the family who carried this base change also carried a silent polymorphism, 843A→G. Genotyping family members for the polymorphic CA microsatellite repeat D17S829 in intron 3 of CTNS allowed for the identification of a mild haplotype (D17S829, allele 3–843G-928A) referred to as haplotype number 3. This haplotype was carried by five family members (I-2, I-3, II-1, II-2, II-3). Various normal haplotypes, with respect to these three markers, were identified in unaffected family members. The second cystinosis allele in patient II-1 was shown by the PCR assay described above to be the common 57-kb deletion. This deletion was presumably inherited from her mother and was not detected in any other available family members. The second, presumed severe, cystinosis allele was not identified in patient II-2. Nevertheless, the two siblings I-2 and I-3 apparently each married an individual heterozygous for cystinosis. The haplotype segregation pattern in the family is depicted in Figure 6, along with leukocyte cystine values.

Pedigree of case 3 (patient II-1) and case 4 (patient II-2). Haplotypes are given to the left of each patient symbol and are numbered using fine type. Haplotype 1, depicted by an empty bar, is normal. Haplotype 2, depicted by a solid bar, carries an unidentified presumably severe mutation in CTNS. Haplotype 3 is the D17S829 (3)-928A-843G haplotype associated with mild disease. Haplotype 4 is another normal haplotype. The Δ haplotype carries the 57-kb deletion involving CTNS. Leukocyte cystine values in nmol half-cystine/mg protein are given beneath each patient symbol.
Northern blots.
RNA was available from fibroblasts of patients 1, 2, and 4 as well as from two normal controls and two cystinosis patients homozygous for the 57-kb deletion in CTNS. Patient 1 showed a small amount of CTNS mRNA (Fig. 7A), 10.5% of normal using a Fujifilm BAS-1500 phosphoimager, whereas patients 4 and 2 exhibited normal or nearly normal levels (Fig. 7B). Patients homozygous for the 57-kb deletion in CTNS had no CTNS mRNA expression in their cultured fibroblasts.

Northern blots of patient CTNS mRNA. A, lanes 1 and 5, normal control fibroblast RNA . Lanes 2 and 4, RNA from 57-kb deletion patients. Lane 3, fibroblast RNA from case 1. Loading can be gauged by the abundance of β-actin mRNA. B, lane 1, normal control fibroblast RNA . Lane 2, RNA from fibroblasts of case 4. Lane 3, RNA from fibroblasts of case 2.
DISCUSSION
The identification of CTNS mutations in four patients with ocular nonnephropathic cystinosis (Table 1) allows for several important conclusions. First, the results confirm that ocular cystinosis is allelic with infantile nephropathic cystinosis. In addition, our findings promote the guarded generalization that ocular cystinosis patients are heterozygous for one severe mutation (e.g. 545delTCCTT in case 1 and the 57-kb deletion in cases 2 and 3) and one mild mutation (e.g. IVS10–3 C→G in case 1 and 928 G→A in cases 2–4). The severe mutations are known to be debilitating because they occur in patients with classic nephropathic cystinosis. The 545delTCCTT has been found in the hemizygous state in a nephropathic cystinosis cell line (28) and the 57-kb deletion in the homozygous state produces classic severe cystinosis (27, 28).
The 928 G→A and splicing mutations are inferred to be mild because they occur in patients with ocular cystinosis. However, additional findings support their mild nature. Although no cystinosin determinations were available, the 928 G→A mutation yields large quantities of CTNS mRNA even in combination with a CTNS deletion allele (case 2, lane 3, Fig. 7B). Moreover, cystinosis heterozygotes bearing 928 G→A plus a normal CTNS allele, i.e. patients I-2, I-3, and II-3 in Figure 6, had leukocyte cystine values that were normal (0.10 nmol half-cystine/mg protein) or only slightly above normal (0.23 and 0.29 nmol half-cystine/mg protein), indicating minimal impairment of lysosomal membrane cystine transport. Finally, whereas the seven previously identified missense mutations causing nephropathic cystinosis have been located either within a predicted transmembrane domain of cystinosin or one amino acid outside of a transmembrane domain (27, 28), the G197R mutation is nine amino acids away from the nearest transmembrane domain. The 928 G→A change should be considered a mutation rather than a polymorphism because it segregated with the disease in cases 3 and 4 and was not present in 55 other sequenced alleles.
The mild nature of the IVS10–3 C→G mutation is supported by several pieces of evidence. First, the maternal mutation, 545delTCCTT, results in termination at codon 73 and is expected to contribute no cystinosin. Second, the IVS10–3 C→G mutation clearly produces some normal mRNA that gives rise to cDNA (lower band, Fig. 4B). In the fibroblasts of case 1, some residual CTNS mRNA (approximately 10% of normal) was apparent (Fig. 7), and the level of expression may be considerably higher in this patient's kidney. Splicing mutations, in general, can be mild by virtue of some degree of normal mRNA processing. Splicing defects caused by mutations in the −3 position of a splice acceptor site are rare; only three have been mentioned in recent publications on the subject (35, 37).
Perhaps the most intriguing question broached by the molecular analysis of ocular cystinosis is why the kidney is spared in these patients. For cases 2–4, one explanation is straightforward. The 928 G→A mutation allows for significant residual activity of cystinosin, consistent with the high level of CTNS mRNA expression in fibroblasts (Fig. 7) and with the relatively low leukocyte cystine values in these patients, i.e. 1.2 and 2.3 nmol half-cystine/mg protein. The cystinosin activity appears sufficient to prevent kidney damage but not enough to spare the cornea, whose lysosomes produce enough cystine to saturate the endogenous cystine carrier and form crystals.
This explanation fails to hold for case 1. First, the residual CTNS mRNA, 10–15% of normal (Fig. 7), and lysosomal cystine countertransport, on average 19% of normal (32), is relatively low. Second, the leukocyte and fibroblast cystine levels are in the range for classic nephropathic cystinosis patients. Clearly, if expression of CTNS is the same in this patient's kidney and leukocytes, this patient should have the same serious kidney disease as other patients with his leukocyte cystine levels. We propose that the sparing of the kidney in case 1 relates to his specific CTNS mutation, the IVS10–3 C→G, affecting splicing. In particular, we suggest that the tissue-specific expression of splicing factors that presumably occurs in all humans allows for increased use of the mutant splice acceptor site in the kidney compared with other tissues in which the cryptic splice site is preferred. This may be because kidney expression of a splicing factor is increased quantitatively or decreased in quality, i.e. it has less specificity in its requirements for acceptor site recognition. It has been previously demonstrated that variable expression of the splicing factor A1 influences the alternative splicing of cystic fibrosis gene (CFTR) splicing mutants (38). Furthermore, any residual normally spliced CTNS mRNA in kidney cells may undergo enhanced expression because the endogenous level of CTNS mRNA is normally increased in kidney compared with other tissues (27). Note that no such increased CTNS expression is possible in the kidney of classic nephropathic cystinosis patients, because their two severe mutations allow no normal CTNS mRNA production.
A final speculation concerns the reason for the high expression of CTNS mRNA in cases 2 and 4. Case 2 has only one functional allele (the one bearing the 928 G→A mutation), which produces a significant fraction of the normal contingent of CTNS mRNA (Fig. 7B). It may be that reduced cystinosin production with consequent elevation in lysosomal cystine results in induction of CTNS at the transcriptional level, accounting for the high level of its mRNA. A similar induction may occur for both the 928 G→A allele and the unidentified allele of patient 4, resulting in a supranormal level of CTNS mRNA (Fig. 7B). Although regulatory activity involving an integral lysosomal membrane protein is not generally considered to exist, a TSH-responsive tyrosine transport system has been described in the lysosomal membrane of rat thyroid (FRTL-5) cells in culture (39). Mutant CTNS alleles may serve as reagents for future investigations into the regulation of cystinosin production.
Abbreviations
- SSCP:
-
single-stranded conformational polymorphism
- RT-PCR:
-
reverse-transcription polymerase chain reaction
References
Gahl WA, Schneider JA, Aula P 1995 Lysosomal transport disorders: cystinosis and sialic acid storage disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease, 7th Ed. McGraw-Hill, New York, 3763–3797.
Gahl WA 1986 Cystinosis coming of age. Adv Pediatr 33: 95–126.
Gahl WA, Kaiser-Kupfer MI 1987 Complications of nephropathic cystinosis after renal failure. Pediatr Nephrol 1: 260–268.
Gahl WA, Dalakas M, Charnas L, Chen KTK, Pezeshkpour GH, Kuwabara T, Davis SL, Chesney RW, Fink J, Hutchison HT 1988 Myopathy and cystine storage in muscles in patients with nephropathic cystinosis. N Engl J Med 319: 1461–1464.
Charnas LR, Luciano CA, Dalakas M, Gilliat RW, Bernardini I, Ishak K, Swik VA, Fraker D, Brushart TA, Gahl WA 1994 Distal vacuolar myopathy in nephropathic cystinosis. Ann Neurol 35: 181–188.
Sonies BC, Ekman EF, Andersson HC, Adamson MD, Kaler SG, Markello TC, Gahl WA 1990 Swallowing dysfunction in nephropathic cystinosis. N Engl J Med 323: 565–570.
Fivush B, Green OC, Porter CC, Balfe JW, O'Regan S, Gahl WA 1987 Pancreatic endocrine insufficiency in post-transplant cystinosis. Am J Dis Child 141: 1087–1089.
Fivush B, Flick JA, Gahl WA 1988 Pancreatic exocrine insufficiency in a patient with nephropathic cystinosis. J Pediatr 112: 49–51.
Ehrich JHHH, Stoeppler L, Offner G, Brodehl J 1979 Evidence for cerebral involvement in nephropathic cystinosis. Neuropaediatrie 10: 128–137.
Fink JK, Brouwers P, Barton N, Malekzadeh MH, Sato S, Hill S, Cohen WE, Fivush B, Gahl WA 1989 Neurologic complications in longstanding nephropathic cystinosis. Arch Neurol 46: 543–548.
Chik CL, Friedman A, Merriam GR, Gahl WA 1993 Pituitary-testicular function in nephropathic cystinosis. Ann Intern Med 119: 568–575.
Kaiser-Kupfer MI, Caruso RC, Minckler DS, Gahl WA 1986 Long-term ocular manifestations in nephropathic cystinosis post-renal transplantation. Arch Ophthal 104: 706–711.
Thoene J, Oshima R, Crawhall J, Olson D, Schneider J 1976 Cystinosis: intracellular cystine depletion by aminothiols in vitro and vivo. J Clin Invest 58: 180–189.
Gahl WA, Reed GF, Thoene JG, Schulman JD, Rizzo WB, Jonas AJ, Denman DW, Schlesselman JJ, Corden BJ, Schneider JA 1987 Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med 316: 971–977.
Markello TC, Bernardini IM, Gahl WA 1993 Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med 328: 1157–1162.
Kimonis VE, Troendle J, Rose SR, Yang ML, Markello TC, Gahl WA 1995 Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab 80: 3257–3261.
Gahl WA, Charnas L, Markello TC, Bernardini I, Ishak KG, Dalakas MC 1992 Parenchymal organ cystine depletion with long term cysteamine therapy. Biochem Med Metab Biol 48: 275–285.
Kaiser-Kupfer MI, Fujikawa L, Kuwabara T, Jain S, Gahl WA 1987 Removal of corneal crystals by topical cysteamine in nephropathic cystinosis. N Engl J Med 316: 775–779.
Kaiser-Kupfer MI, Gazzo MA, Datiles MB, Caruso RC, Kuehl EM, Gahl WA 1990 A randomized placebo-controlled trial of cysteamine eye drops in nephropathic cystinosis. Arch Ophthalmol 108: 689–693.
Cogan DG, Kuwabara T, Kinoshita J, Sheehan L, Merola L 1957 Cystinosis in an adult. JAMA 164: 394–396.
Pellet OL, Smith ML, Greene AA, Schneider JA 1988 Lack of complementation in somatic cell hybrids between fibroblasts from patients with different forms of cystinosis. Proc Natl Acad Sci USA 85: 3531–3534.
Gahl WA, Tietze F, Bashan N, Steinherz R, Schulman JD 1982 Defective cystine exodus from isolated lysosome-rich fractions of cystinotic leucocytes. J Biol Chem 257: 9570–9575.
Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD 1982 Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217: 1263–1265.
Jonas AJ, Smith ML, Schneider JA 1982 ATP-dependent lysosomal cystine efflux is defective in cystinosis. J Biol Chem 257: 13185–13188.
Gahl WA, Tietze F, Bashan N, Bernardini I, Schulman JD 1983 Characteristics of cystine counter-transport in normal and cystinotic lysosome-rich leucocyte granular fractions. Biochem J 216: 393–400.
The Cystinosis Collaborative Research Group 1995 Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat Genet 10: 246–248.
Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van't Hoff W, Antignac C 1998 A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 18: 319–324.
Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, Guo J, Thoene J, Gahl WA 1998 CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet 63: 1352–1362.
Anikster Y, Lucero C, Touchman JW, Huizing M, McDowell G, Shotelersuk V, Green ED, Gahl WA 1999 Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS). Mol Genet Metab 66: 111–116.
Oshima RG, Willis RC, Furlong CE, Schneider JA 1974 Binding assays for amino acids. J Biol Chem 249: 6033–6039.
Charnas LR, Bernardini I, Rader D, Hoeg JM, Gahl WA 1991 Clinical and laboratory findings in the oculocerebrorenal syndrome of Lowe, with special reference to growth and renal function. N Engl J Med 324: 1318–1325.
Gahl WA, Tietze F 1987 Lysosomal cystine transport in cystinosis variants and their parents. Pediatr Res 21: 193–196.
Wong C, Antonarakis SE, Goff SC, Orkin SH, Forget BG, Nathan DG, Giardina PJ, Kazazian HH 1989 Beta-thalassemia due to two novel nucleotide substitutions in consensus acceptor splice sequences of the beta-globin gene. Blood 73: 914–918.
Ieiri T, Cochaux P, Targovnik HM, Suzuki M, Shimoda S, Perret J, Vassart G 1991 A 3′ splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest 88: 1901–1905.
Holzl B, Huber R, Paulweber B, Patsch JR, Sandhofer F 1994 Lipoprotein lipase deficiency due to a 3′ splice site mutation in intron 6 of the lipoprotein lipase gene. J Lipid Res 35: 2161–2169.
Jaruzelska J, Abadie V, d'Aubenton-Carafa Y, Brody E, Munnich A, Marie J 1995 In vitro splicing deficiency induced by a C to T mutation at position −3 in the intron 10 acceptor site of the phenylalanine hydroxylase gene in a patient with phenylketonuria. J Biol Chem 270: 20370–20375.
Krawczak M, Reiss J, Cooper DN 1992 The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90: 41–54.
Chiba-Falek O, Nissim-Rafinia M, Kerem B 1998 Regulation of CFTR alternative splicing in vivo by overexpression of cellular and viral splicing factors. Am J Hum Genet 63: A356–A359
Harper GS, Kohn LD, Bernardini I, Bernar J, Tietze F, Andersson H, Gahl WA 1988 Thyrotropin stimulates lysosomal tyrosine transport in rat FRTL-5 thyroid cells. J Biol Chem 263: 9320–9325.
Acknowledgements
Dr. Yair Anikster is a Howard Hughes Medical Institute Physician Postdoctoral Fellow. The authors thank Mr. Ernest Kuehl of the National Eye Institute for superb ophthalmic photography. Cases 3 and 4 were astutely diagnosed and kindly referred by Dr. Walter Beebe of Dallas, Texas.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Anikster, Y., Lucero, C., Guo, J. et al. Ocular Nonnephropathic Cystinosis: Clinical, Biochemical, and Molecular Correlations. Pediatr Res 47, 17 (2000). https://doi.org/10.1203/00006450-200001000-00007
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200001000-00007
This article is cited by
-
Molecular characterization of CTNS mutations in Tunisian patients with ocular cystinosis
Diagnostic Pathology (2022)
-
A comparison of immediate release and delayed release cysteamine in 17 patients with nephropathic cystinosis
Orphanet Journal of Rare Diseases (2021)
-
Slow progression of renal failure in a child with infantile cystinosis
CEN Case Reports (2018)
-
Latest Clinical Approaches in the Ocular Management of Cystinosis: A Review of Current Practice and Opinion from the Ophthalmology Cystinosis Forum
Ophthalmology and Therapy (2018)
-
Mutation analysis of the CTNS gene in Iranian patients with infantile nephropathic cystinosis: identification of two novel mutations
Human Genome Variation (2017)
