
Abstract
Glucocorticoids (GCs) are essential for many aspects of normal brain development. However, there is growing evidence from a number of species that exposure of the fetal brain to excess GC, at critical stages of development, can have life-long effects on behavior and neuroendocrine function. The hypothalamo-pituitary-adrenal axis, which is central to the integration of the individual's endocrine and behavioral response to stress, appears highly sensitive to excess GC exposure during development. A number of animal studies have shown that exposure to synthetic GCs in utero results in adult offspring that exhibit hyperactivity of the hypothalamo-pituitary-adrenal axis. This will have a long-term impact on health, inasmuch as increased life-long exposure to endogenous GC has been linked to the premature onset of diseases associated with aging. The mechanisms involved in the permanent programming of hypothalamo-pituitary-adrenal function and behavior are not well understood. Synthetic GCs are used extensively to promote pulmonary maturation in fetuses at risk of being delivered before term. Therefore, it is important that we understand the potential long-term consequences of prenatal GC exposure on brain development as well as the underlying mechanisms involved. This review will explore the current state of knowledge in this rapidly expanding field.
Similar content being viewed by others

Main
GCs (cortisol in humans and most mammals, and corticosterone in rats, mice, and other lower vertebrates) are essential for normal brain development. They exert a wide spectrum of effects in most regions of the developing brain, ranging from subcellular reorganization to neuron–neuron and neuron–glial interaction (1, 2). However, sustained elevation in, or removal of these hormones from, the fetal brain is detrimental to these processes and can permanently modify the structure and function of the brain (3, 4).
The pioneering work of Liggins and Howie (5) in the early 1970s has led to the widespread use of synthetic GCs to treat fetuses at risk of preterm delivery. Preterm delivery occurs in approximately 7–10% of all births in North America and is responsible for about 75% of neonatal deaths (6). In these cases, neonatal morbidity is high in surviving preterm infants, and complications such as respiratory distress syndrome, intraventricular hemorrhage, and necrotizing enterocolitis are common (6). Prenatal GC therapy reduces the frequency of the complications associated with preterm delivery (6). As a result, in 1994 the National Institutes of Health Consensus Development Conference recommended antenatal treatment of all women at risk of preterm delivery, between 24 and 34 wk of gestation, with GCs (6). However, the consensus report highlighted the requirement for further research into the effects of antenatal GCs on brain development and function after birth. This review will explore the current state of knowledge in this area. Although there is a growing literature on the impact of prenatal GC exposure on cardiovascular and metabolic function after birth [for recent review, see Dodic et al. (7)], this aspect of programming will not be considered in detail in this review.
In addition to use in cases of preterm labor, synthetic GCs are also administered to pregnant women in other clinical situations, including congenital adrenal hyperplasia. Often these treatments occur in early pregnancy and in some cases throughout gestation. There is currently very little information about the impact of such treatment regimens on brain and neuroendocrine development. However, some of the long-term effects discussed in this review may also occur in these clinical situations.
DEVELOPMENT OF BRAIN AND NEUROENDOCRINE SYSTEMS
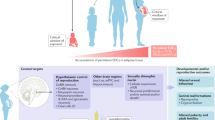
Within the developing brain, the limbic system (primarily the hippocampus) is particularly sensitive to endogenous and exogenous GCs during development (8–10). The hippocampus has a myriad of complex functions within the brain. These include cognition, behavior, memory, coordination of autonomic activity, and regulation of a number of endocrine systems (10–12). Given the wide spectrum of regulatory roles, programming of limbic function during development will have a profound impact in postnatal and adult life (Fig. 1).

Diagrammatic representation of the routes by which prenatal GC exposure programs adult behavior and neuroendocrine function. The fetal limbic system (primarily the hippocampus), hypothalamus, and anterior pituitary express high concentrations of corticosteroid receptors, and are sensitive to GCs. Exposure to exogenous GC at this time will alter development and subsequent function of both the limbic system and the HPA axis. The hippocampus regulates HPA function, and endogenous GCs (the end product of HPA activation) modify many aspects of limbic function. The gray arrows indicate this close functional association. In the periphery, the overall effect of programming during development will be altered exposure to endogenous GC throughout life. Increased exposure will predispose to a number of neurologic, metabolic, and cardiovascular diseases, whereas reduced exposure may act to protect against these diseases. See text for further details.
It has been known for four decades that HPA function can be permanently programmed during development (13), although the mechanisms have remained unclear. The HPA axis is modulated by efferent outflow from the limbic system [for review, see Jacobson and Sapolsky (12) and Herman et al. (14)] (Fig. 2). GCs act at multiple loci within the body to maintain homeostasis, but also act in the CNS to modify behavior and learning [for review, see Munck et al. (15) and Lupien and McEwen (16)]. Modification of systems that regulate HPA function during development will have major consequences on behavior and other higher center function, as well as on the long-term health of the individual (Fig 1).

A schematic representation of the HPA axis. The hypothalamic PVN controls pituitary-adrenocortical activity [for review, see Antoni (29) and Whitnall (30)]. The limbic system, primarily via the hippocampus, forms a major inhibitory input to the PVN (12). There are few direct connections between the hippocampus and the PVN, but many indirect links via the bed nucleus of the stria terminalis and the ventromedial hypothalamus (12, 14). Parvocellular neurons in the PVN synthesize CRH and AVP, which are then released into the hypophysial portal circulation (31). CRH and AVP stimulate ACTH synthesis and release from corticotrophs in the anterior pituitary gland (31). ACTH then initiates the synthesis and secretion of cortisol from the adrenal cortex (32). Because of the damaging effects of extended tissue exposure to GCs, the HPA axis is tightly regulated (32, 33). GCs feed back, via GR and MR, at several sites to inhibit further HPA activity (10, 12, 34, 35). Synthetic GCs bind predominantly to the GR (36), and the effects on development after prenatal synthetic GC administration are likely mediated at the level of this receptor. POMC, pro-opiomelanocortin.
The timing of maturation of the HPA axis relative to birth is highly species specific and is closely linked to landmarks of brain development (17, 18). In animals that give birth to mature young (sheep, guinea pigs, and primates) maximal brain growth and a large proportion of neuroendocrine maturation (including corticosteroid receptor development) takes place in utero (19, 20). In contrast, in species that give birth to immature young (rats, rabbits, and mice), much neuroendocrine development occurs in the postnatal period (21). Therefore, maternal GC treatment in late gestation will impact on different stages of brain and HPA development depending on the species studied. Another important consideration when extrapolating among different studies and species is that of receptor sensitivity. In this connection, mice and rats are corticosensitive (high receptor affinity for GCs) compared with other species, such as guinea pigs and primates, which are considered corticoresistant (22).
Under normal circumstances, and in all species studied, access of maternal endogenous GCs (cortisol and corticosterone) to the fetus is low (23). This is related, in part, to the expression of 11β-HSD in the placenta (24) This area has previously been reviewed in detail (25, 26). Briefly, 11β-HSD interconverts cortisol and corticosterone to inactive products (cortisone, 11-dehydrocorticosterone). There are two known isoforms, 11β-HSD type 1, which is bidirectional, and type 2, which is unidirectional (cortisol to cortisone) (24). The efficiency of placental 11β-HSD2 varies considerably among species (24, 27, 28); however, it is generally accepted that placental 11β-HSD2 is of primary importance in excluding maternal GC from the fetus (24, 25). 11β-HSD2 has a low affinity for synthetic GCs (25), and so dexamethasone and betamethasone pass rapidly from mother to fetus.
This review will focus on the impact of synthetic GCs in the developing brain and resultant changes in neuroendocrine function and behavior. However, studies that have used endogenous GCs will be cited when no information using synthetic GCs is available.
PRENATAL GC AND ADULT HPA FUNCTION
In the rat, fetal exposure to synthetic GC (0.1 mg/kg) during the last week of gestation resulted in elevated basal plasma corticosterone levels in adult male offspring (37). This increase in corticosterone was associated with an increase in adult blood pressure. There was also a dexamethasone-induced reduction in birth weight, but no effect on gestation length (37). In another study, maternal dexamethasone treatment (0.05 mg/kg) on gestational day 17, 18, and 19 resulted in adult male offspring that mounted a greater corticosterone response to stress (38). In contrast, dexamethasone (0.4 mg/kg) treatment on gestational day 17 and 19 failed to alter basal HPA function in prepubertal offspring. However, there was a significant difference in the ratio of AVP and CRH in the external zone of the median eminence in the young rats (39), indicating that there were subtle long-term effects on HPA regulation. These studies clearly demonstrate that dexamethasone-exposure in utero can program adult HPA function, but that the nature of the modification is dependent on dose and timing of exposure.
Administration of dexamethasone to pregnant guinea pigs on gestational day 50 and 51 (75% of gestation) resulted in prepubertal male, but not female, offspring with dramatically elevated basal plasma cortisol concentrations (40). There was no effect of GC administration on fetal, term, or juvenile body or organ weights, although there was a significant increase in gestation length (40, 41). A single study has been undertaken to establish the long-term effects of fetal exposure to synthetic GCs on HPA function in the primate (42). Pregnant rhesus monkeys were treated daily with dexamethasone (4 × 1.25 mg/kg) commencing on 132 d of gestation. Basal and stress-stimulated cortisol levels were elevated in the offspring (10 mo of age) born to dexamethasone-treated mothers (42). Taken together, these studies demonstrate that short-term fetal exposure to synthetic GCs can program HPA function in species that give birth to neuroanatomically mature young (primates and guinea pigs). To date, no studies have assessed HPA function in school-age children or young adults who were exposed to antenatal GC during fetal life.
In summary, available evidence in animals indicates that prenatal exposure to GCs leads to offspring with elevated HPA activity. There is currently no information as to how programmed changes in pituitary-adrenocortical activity are modified with age. Increased HPA tone throughout life will have a considerable impact on adult health because of elevated tissue exposure to GCs. In humans, elevated concentrations of cortisol have been associated with increased atherosclerosis, cholesterol concentrations, incidence of diabetes, immunosuppression, and cognitive impairment (16, 43, 44). Animal studies have found similar effects of chronically elevated HPA activity (45). Taken together, these data would indicate that chronically elevated HPA function leads to accelerated aging. In contrast, life-long exposure to reduced HPA activity may tend to slow down the onset of age-related diseases.
PRENATAL GC AND ADULT BEHAVIOR
Altered hippocampal development will have considerable effects on adult behavior. This effect may be direct or indirect, via alterations in HPA function (10, 11). In rats, treatment of pregnant dams with corticosterone (endogenous GC) resulted in sex-specific alterations in spontaneous and apomorphine-induced motor activity in adult offspring (46). In adult male offspring exposed to corticosterone as fetuses, exploratory activity was increased, whereas in females, spontaneous locomotion was increased, but there was no effect on exploratory activity (46). In adult female offspring, there was an interaction between apomorphine-induced motor activity and prenatal GC exposure, indicating GC-induced alterations in central dopamine systems (46). Daily dexamethasone treatment (0.1 mg/kg) during the last week of gestation had significant detrimental effects on sexual behavior, such that male offspring became demasculinized and feminized (47).
In mice, maternal treatment with prednisone resulted in offspring with delayed development of eye opening, lifting, walking, and gripping, indicating a retardation of muscular and motor development (48). In another series of studies, either single (49) or repeat doses (50) of dexamethasone or betamethasone (0.1–0.2 mg/kg) were administered to pregnant mice during the last week of gestation. After a single injection of GC on gestational day 14, there were specific differences in anxiety, memory, and socialization when compared with control mice. However, there was no effect of GC on sensory, motor, motivation, and learning performance (49). Together, these studies suggest that prenatal GC exposure has significant but subtle effects on the behavior of adult rodent offspring and that these effects are sex specific.
In rhesus monkeys, there was no effect of prenatal dexamethasone treatment (1.25 mg/kg per day for 4 d) at approximately 0.75 gestation on physical development or locomotor behavior in young primates, although there were dramatic differences in hippocampal structure (42). In humans, a number of studies have been undertaken to establish the long-term effects of prenatal GC exposure on learning, behavior, and intelligence. However, these have been undertaken in children who were born before term (51–54), and therefore were already at risk of delayed neurologic development (55–58). In these studies, there was no overall neurologic delay or impairment in children who were exposed to synthetic GCs in late gestation (51–54). However, in 6-y-old children, antenatal GC exposure was associated with subtle effects on neurologic function, including reduced visual closure and visual memory (53). A single study has considered the impact of GC on cognitive and behavioral development in children who were exposed to repeated antenatal GC treatment but not born before term (59). Children exposed to dexamethasone in early pregnancy, because of increased risk of congenital adrenal hyperplasia, showed significant increases in emotionality, unsociability, avoidance, and behavioral problems. Although no cognitive changes were noted, examination was performed at an age (6 mo to 5.5 y) when differences in cognitive function are difficult to discern. A recent study has shown that repeated courses of GC (each course; 2 × 11.4 mg, 12 to 24 h apart) in pregnant women at increased risk of delivering before term resulted in children with reduced head circumference (60). Further studies are required to establish the impact of repeated antenatal GC treatment, in late gestation, on behavioral outcome in children born at term.
MECHANISMS OF PROGRAMMING
Developmental and functional considerations.
The impact of fetal GC exposure is dependent on the expression of corticosteroid receptors in the developing brain at the time of exposure. A number of studies have shown that levels of GR and MR in the rat brain are low through gestation, but increase rapidly after birth (61–64). This would be consistent with the postnatal nature of brain and HPA development in this species (18, 21). However, more detailed analysis has revealed that there is a distinct ontogenic pattern for GR and MR in the fetal rat brain (65–67). GR mRNA is present in the anterior hypothalamus, hippocampus, and pituitary by gestational day 13, and mRNA levels increase around term (65, 67). In contrast, MR mRNA is not present in the hippocampus until gestational day 16 and the hypothalamus until day 17 (67). It is unclear how fetal levels compare with those in the neonate and adult.
In the fetal guinea pig, GR and MR mRNA are present in the cortex and all regions of the hippocampus and dentate gyrus by gestational day 40 and then throughout the remainder of gestation (20) (Fig. 3). Between gestational day 40 and gestational day 50, there is a dramatic increase in GR mRNA levels, but a decrease in MR mRNA abundance (20), indicating differential developmental regulation of the two receptors in the hippocampus. Hippocampal GR mRNA levels increase to a peak near term, but there is little change in MR mRNA, which remains low. In contrast, in the PVN, GR mRNA levels are higher at gestational day 40 than at any other stage of fetal or postnatal life. There is a dramatic decrease (50%) in GR mRNA near term (20). This likely represents a decrease in GC feedback in the PVN, allowing ACTH and cortisol to increase simultaneously at this time (20, 68). A similar reduction in GR mRNA in the PVN has been noted in the fetal sheep near term (69). In summary, these studies indicate that the development of GR and MR expression is highly species specific. Differences in receptor number likely account for the inconsistent outcomes observed after varied treatment regimens, as well as the differences observed between species.

Color-enhanced image illustrating the expression of GR and MR mRNA in coronal sections of the fetal guinea pig brain at 50 d gestation (term, 70 d). Receptor mRNA was determined by in situ hybridization using 35S-labeled oligonucleotide probes specific for GR and MR. GR mRNA is present at high levels in the CA1–CA4 fields of the hippocampus, the dentate gyrus (DG), PVN, and amygdala (AMG). Lower concentrations of GR mRNA are present in other brain regions. In contrast, MR mRNA is confined almost exclusively to the limbic system. Red, high expression; yellow, moderate expression; green, low expression; blue, no expression.
In the brain, MR has a higher affinity than GR for GC, and at basal concentrations of cortisol, MR is occupied while GR remain largely unoccupied (10). However, during periods of elevated plasma cortisol (i.e. during stress), there is increased occupation of the GR [for review, see Dallman et al. (35)]. It has been proposed that the hippocampal MR is primarily involved in feedback regulation during basal secretion, whereas the GR becomes important during periods of increased GC secretion (35), although this remains somewhat controversial (10). Synthetic GCs bind predominantly to the GR (36), and the effects on development after prenatal GC administration are likely mediated at the level of this receptor. In addition to binding to classic GR, synthetic GCs such as dexamethasone may also bind to neurosteroid receptors in the brain (70). This would represent a novel route of signal transduction and clearly warrants further investigation.
GCs and central neurotransmitter systems.
The catecholamines epinephrine and norepinephrine stimulate HPA function via α1 adrenergic receptors [for review, see Plotsky et al. (71)]. Catecholaminergic innervation of the parvocellular PVN is derived principally from the caudal medulla (72, 73). Brainstem catecholaminergic systems are also implicated in hippocampal function. Stress increases norepinephrine turnover in the hippocampus and prefrontal cortex (74, 75), and norepinephrine has been shown to modulate hippocampal corticosteroid receptor activity (76).
Dexamethasone-exposure in the last week of gestation results in adult offspring with reduced norepinephrine turnover in the cerebellum and forebrain (77) and reduced norepinephrine content in the hippocampus and neocortex (38). However, longitudinal studies have shown that antenatal GC exposure results in premature maturation of norepinephrine systems in the brainstem, forebrain, and cerebellum (77), and induces overexpression of the norepinephrine transporter (78). Unfortunately, norepinephrine concentrations and turnover and norepinephrine transporter concentrations have not been measured in the hypothalami of offspring that were exposed to GCs as fetuses. No effect of prenatal dexamethasone on adrenergic receptors has been detected (77). Without further measurements in the hypothalamus, it is unclear how dexamethasone-induced modification of adult brain norepinephrine systems relates to the increase in HPA function. However, it has been shown that alterations in hippocampal norepinephrine can induce changes in corticosteroid receptor levels (79) and therefore modify GC negative feedback. Antenatal GCs have also been shown to promote early maturation of dopamine systems in the forebrain (77), and this is likely associated with subtle changes in behaviors noted in these animals.
Ascending serotonergic neurons project directly to the parvocellular PVN and increase activity of CRH-containing neurons (80, 81), stimulating CRH and ACTH secretion into the hypophysial portal blood (82–84). There is also a rich serotonergic innervation of the hippocampus (85). Prenatal exposure to dexamethasone in the last week of gestation leads to male offspring with increased hypothalamic and medullary serotonin concentrations, but reduced hippocampal serotonin turnover (38). Prenatal GC exposure also promoted brainstem serotonin transporter development and resulted in increased transporter activity throughout the life of the animal (86). Measurement of transporter activity was only undertaken in the brainstem, and potential GC-induced changes in the hippocampus and hypothalamus remain to be determined. Alterations in serotonergic transporter function and increased hypothalamic serotonin concentrations are likely responsible, in part, for the elevation of HPA activity observed in adult offspring. The effect of serotonin on HPA function is not confined to the PVN inasmuch as serotonin tonically inhibits the expression of hippocampal MR in adult male rats (87).
GCs and central corticosteroid receptors.
In the rat, dexamethasone (0.1 mg/kg) administered during the last week of gestation resulted in adult male offspring with reduced concentrations of GR mRNA and MR mRNA in the hippocampus (37). There was no difference in GR or MR expression in the hypothalamus or in brainstem structures. Until recently, it was thought that occupation of the hippocampal MR and GR is associated with suppression of HPA activity (12). Therefore, a reduction in expression would be associated with an increase in pituitary-adrenocortical activity. However, this view has recently been challenged, and it has been suggested that occupation of the hippocampal GR can, under certain circumstances, act to potentiate activated HPA function, whereas occupation of the MR suppresses activity (10). The reduction of hippocampal MR mRNA in animals that were exposed to GCs is consistent with increased basal and activated HPA activity (i.e. reduced GC feedback) in this model. However, the functional role of the decrease in hippocampal GR mRNA is less clear, and requires further study.
In the guinea pig, it has recently been demonstrated that prenatal GC exposure, during the phase of rapid brain growth (gestational day 50 and 51), results in juvenile offspring with an altered hippocampal corticosteroid receptor complement (40). This effect is highly sex specific. Male offspring exhibit increased GR mRNA concentrations in the cingulate cortex and hippocampal CA3, whereas females exhibit reduced GR mRNA in the hippocampus (CA3 and CA4), dentate gyrus, and cingulate cortex (40). There was no effect of prenatal GC on concentrations of GR mRNA in the hypothalamus and anterior pituitary. Whether the hippocampal differences are maintained into adulthood remains to be determined.
There are a number of additional, and as yet uninvestigated, levels at which changes in central GC sensitivity may be mediated [for review, see Bamberger et al. (88)]. It is possible that prenatal GCs may have long-term influences on 1) intracellular hormone availability (67, 89, 90), 2) interactions between receptor and heat-shock protein (91), 3) receptor phosphorylation (92), 4) nuclear translocation (93), and 5) DNA binding and interaction (94). Further studies are required to verify this possibility.
Mechanisms of corticosteroid receptor programming.
In both the rat and guinea pig, there are rapid changes in the expression of GR and MR around the time of maximal brain growth (guinea pig gestational day 50; rat postnatal day 8) (18). It is possible that GC exposure can have permanent effects on the trajectory of GR and MR development at this time, such that the hippocampal receptor complement is permanently altered. Very little is known about the mechanisms by which GCs modulate developing GR and MR populations. Likely mechanisms include autoregulation or modification by brain biogenic amines. High concentrations of GC in the brain may act to reduce (autoregulate) corticosteroid receptor expression. This area has been reviewed in detail (88). Briefly, it has been shown that synthetic GCs down-regulate GR mRNA by 50–80% in a variety of cell types (95). Of particular relevance to this discussion, extended exposure of HeLa cells to dexamethasone resulted in irreversible down-regulation of GR mRNA, a consequence of conformational change in the GR promoter (96).
Only one study has investigated the immediate impact of synthetic GCs on hippocampal corticosteroid receptor concentrations in the fetus (41). In the guinea pig, injection of dexamethasone on gestational day 50 and 51 increased GR and MR in the CA1 region of the hippocampus in female fetuses. Day 50 of gestation is a time of rapid brain growth and very low plasma cortisol concentrations in the fetal guinea pig (20, 68). In rats, Kalinyak and colleagues (97) found no change in total brain GR mRNA 6 h after a single dexamethasone injection in pups less than a week of age, and daily injections of corticosterone for 5 d were found to cause only small decreases in hippocampal dexamethasone binding in rat pups (98). Together, these studies would suggest that autoregulation by high doses of synthetic GC does not occur during periods of low adrenocortical activity during development. However, this does not preclude the possibility that autoregulation (i.e. reduced receptor number) by GCs may occur earlier in development, before the animals enter the adrenal hyporesponsive period. It is also unclear whether the effects of the prenatal treatment on corticosteroid receptors observed in young animals will reflect the receptor complement in the adult hippocampus. Emerging evidence from our laboratory would suggest that this may not be the case. In female guinea pig fetuses, dexamethasone exposure increased hippocampal MR and GR mRNA (41), but the same fetal treatment resulted in prepubertal female offspring with reduced hippocampal GR mRNA (40). Currently, there is very little information on the influence of exogenous GC on neurotransmitter activity in the fetal brain or on the impact of neurotransmitter concentrations on corticosteroid receptors in the fetal brain.
Mitchell and colleagues (99) have recently demonstrated that serotonin regulates GR in primary hippocampal cultures derived from fetal rat brain. Serotonin treatment resulted in an increase in GR binding, and this effect was mediated via high-affinity serotonin (5-HT2) receptors. Furthermore, the effect was permanent, inasmuch as increased levels of GR were maintained for 60 d, in the absence of further serotonin exposure (99). This group also demonstrated that serotonin was responsible for the up-regulation of hippocampal GR, previously demonstrated in adult rats that were handled as neonates (100, 101). Handling has been shown to increase thyroid activity in the neonate (101, 102). Furthermore, neonatal thyroid hormone administration mimics the effects of handling on GR binding in adulthood (103). Subsequent studies demonstrated that both neonatal handling and thyroid hormone administration resulted in increased serotonin turnover in the hippocampus (102). As a result, it has been suggested that the ascending serotonin neurons are stimulated by thyroid hormones to increase hippocampal serotonin and thence GR expression (101). Prenatal GC exposure has also been shown to advance maturation of the serotonin transporter system in the developing rat brain (86). In the fetal guinea pig, exposure to dexamethasone resulted in an increase in fetal thyroid hormone and an up-regulation of GR mRNA (41). Together these data indicate that an increase in central serotonin concentrations may mediate the dexamethasone-induced increases in hippocampal GR mRNA in the fetus (41), although further work is required to confirm this possibility.
PRENATAL GC AND BRAIN STRUCTURE
The effects of GC are not restricted to central changes in neurotransmitter concentrations and receptor numbers, but may also include modification of brain structure. Several studies in young animals and adults have demonstrated that stress and increased GC can have a major impact on hippocampal structure [for review, see De Kloet et al. (10)]. Perhaps the most striking of these are the observations that stressful experience or exposure to chronically elevated GC results in significant reductions in hippocampal volume (104–107).
An elegant series of studies performed in the rhesus monkey have considered the impact of short periods of fetal GC exposure during late gestation on structural development of the limbic system (42, 108). The effect of single injections of dexamethasone (0.5, 5, and 10 mg/kg) at 132 d gestation (term, 185 d) was assessed on gestational day 135. There was considerable dose-dependent neuronal degeneration in the CA1–CA3 hippocampal pyramidal neurons, shrinkage and condensation of neuron soma and dendritic branches, depletion of the number of neurons, and disintegration of mossy fiber endings in the zona lucidum (108). Fetuses receiving multiple injections of lower doses (0.125 × 4, 1.25 × 4, and 2.5 mg/kg × 4) showed more severe damage than those receiving a single larger injection. This would be more analogous to the treatment regimen in pregnant women, and the lower dose would be similar to that prescribed for suspected preterm labor (6).
Another group of fetuses was studied 30 d after exposure, and although the acute neurodegenerative changes seen in the brains of the day 135 fetuses had subsided, the size of the perikaryonic soma of the pyramidal neurons was small, and their dendritic branches with the mossy fiber endings in the CA3 regions were poorly developed compared with normal age-matched controls (108). In the dentate gyrus, the density of the granular neurons was less, and the individual sizes of the neurons were smaller. Overall, the number of hippocampal neurons at day 162 was reduced approximately 30% in the dexamethasone-treated fetuses, and the size of the whole hippocampal formation was reduced (108). In a further experiment involving dexamethasone-treated fetal monkeys, the hippocampal volume was analyzed at 20 mo of age by magnetic resonance imaging. This revealed an approximate 30% reduction in hippocampal volume in offspring exposed to dexamethasone (4 × 1.25 mg/kg) at day 132 of gestation (42).
Human and animal studies have demonstrated that morphologic changes in the hippocampus are associated with a number of functional consequences (104). Bremner et al. (106) showed that male combat veterans with posttraumatic stress disorder had reduced magnetic resonance imaging–derived right-sided hippocampal volume compared with control subjects, and Sheline et al. (105) reported that certain aspects of their memory deficit were correlated with hippocampal volume. More recent studies have demonstrated a reduction in hippocampal volume in women victimized by childhood sexual abuse and that the severity of psychiatric complications was correlated to reduction in hippocampal volume (107). It is therefore likely that GC-induced changes in hippocampal structure during development have long-term behavioral and neuroendocrine consequences.
The mechanisms by which GC-induced damage to the hippocampus takes place are not well understood. It is generally accepted that GCs promote differentiation over proliferation (10, 109). Animal studies have demonstrated that GCs interfere with normal rates of cell birth and death that occur during development (2). However, it is also known that GC exposure can indirectly damage mature neurons (110). It has been shown that GCs block the uptake of glucose into neurons (111). Under resting conditions, GCs do not reduce energy metabolism sufficiently to kill neurons; however, during situations such as ischemia or glucose insufficiency, GCs decrease energy metabolism, making neurons more vulnerable to challenge (112). It is thought that glutamate, which is released during episodes of hypoxia and hypoglycemia, is responsible for inducing damage in compromised neurons (110). GCs have also been shown to increase extracellular glutamate concentrations in the hippocampus by preventing glutamate reuptake, and therefore further exacerbating damage (112). This damage is consistent with that described in fetal primates exposed to GC.
Other aspects of CNS development are also affected by prenatal GC exposure. A recent study in sheep has shown that repeated fetal exposure to GCs delays myelination of optic axons (113). In rats, fetal dexamethasone exposure tends to promote the replacement of neurons with glia in the forebrain (114), and has sustained effects on the formation of polyamines (115). The latter are major regulators of neural cell replication and differentiation. GCs also regulate the expression of the neuronal cell adhesion molecule (116), which is important in maturation and synaptic stabilization (117). A recent study has shown that a single low dose of dexamethasone (0.05 mg/kg) can cause profound perturbation of nuclear transcription factors in the fetal rat brain (118). Indeed, low doses of dexamethasone caused changes in fetal brain c-fos gene expression and AP-1 binding proteins of a magnitude the same as or larger than those associated with GC-induced teratogenesis (119), suggesting that the brain is especially vulnerable to GC exposure in late gestation.
Neurotrophic factors are essential for development of interneuronal and neuronal-glial interactions (120, 121). Indeed, both stress and GCs affect the expression of neurotrophic factors in limbic and hypothalamic structures in the adult brain (122–125). Several neurotrophic factors exist in the developing brain, although virtually nothing is known about the functional regulation of neurotrophic factors during development (121, 126). It is highly likely that neurotrophic factors play a role in prenatal GC programming of hippocampal and hypothalamic function by modifying neuron–neuron and neuron–glial interaction, within and between these structures.
It is possible that compromise (i.e. reduction of pyramidal neurons or alterations in axonal or dendritic processes and synaptogenesis) in the developing hippocampus may decrease the age at which hippocampal deficits are first noted. Prenatally programmed increases in HPA function may exacerbate this hippocampal deficit (104, 110, 127) and, in turn, lead to further increases in HPA function (because of reduced GC negative feedback). In this connection, aging humans tend to undergo a decrease in HPA resiliency (ability to switch off the HPA response), and it has been suggested that this deficit is linked to life-long exposure to GCs [for review, see Seeman and Robbins (33)]. For these reasons, follow-up studies in humans and animal models, to investigate the impact of prenatal GC exposure, may fail to observe significant hippocampal deficits until adulthood or early old age.
SUMMARY AND CONCLUSIONS
In most of the studies cited in this review, the doses and regimen of synthetic GC administration were not dissimilar from those administered to pregnant women at risk of delivering before term. In a recent study, a very low dose of GCs in late gestation has been shown to have dramatic effects on transcription factor expression in the developing brain (118). An important consideration when extrapolating among different studies and species is that of receptor sensitivity. In this connection, mice and rats are corticosensitive (i.e. high receptor affinity for GCs) compared with guinea pigs and primates (including humans), which are considered corticoresistant (22). Current evidence indicates that prenatal GC exposure can program brain and neuroendocrine function in both corticosensitive and corticoresistant animals. The magnitude of effect will also depend on the corticosteroid receptor complement within the brain at the time of treatment as well as the sex of the fetus. This is highly species and brain region specific. Recent studies have indicated that maturation of the GR and MR systems is advanced in species in which rapid brain development occurs in utero (guinea pigs and sheep), and this is likely also the case in primates.
The preceding evidence has led many investigators to suggest that alterations in hippocampal corticosteroid receptor regulation are responsible for the programming of HPA and alterations in adult behavior (Fig. 4). However, fundamental questions that remain include the following:1) At which point in development are these receptor systems susceptible to programming? 2) In the fetus, are the effects of excess GCs mediated directly at the hippocampal receptor or via indirect routes (i.e. changes in neurotransmitter systems or structural interactions)? 3) Does programming of receptor number occur solely during early development or do other changes in early life (i.e. permanent changes in neurotransmitter systems) lead to persistent alterations in GR and MR regulation in adulthood? 4) What proportion of the programming effect is not associated with changes in corticosteroid receptors, but rather with changes in other prenatally programmed systems? 5) What are the mechanisms involved in the sex-specificity of HPA programming? Although not considered in detail in this review, it should also be noted that there are likely other indirect routes by which prenatal GC exposure alters fetal brain development. For example, treatment with antenatal GCs results in an increase in placental CRH production in humans (128, 129). Placental CRH may enter the fetus and activate the HPA axis; however, it may also have direct effects on blood flow within the placenta (130, 131). Further research is required to better delineate the complex mechanisms by which GCs affect the developing fetus.

Diagram illustrating the potential routes by which prenatal GC exposure leads to alterations in behavior and HPA activity in adulthood. During development, fetal exposure to GCs directly affects 1) development and subsequent function of neurotransmitter systems (and their transporter mechanisms) in the brainstem, 2) development of corticosteroid receptor expression and structural components in the hippocampus, and 3) development and subsequent function of parvocellular neurons (CRH/AVP). Because the brainstem neurotransmitter systems project directly to the hippocampus and PVN, GC-induced changes in neurotransmitter and transporter systems will indirectly affect function of the hippocampus and PVN. Also, given the important role of the hippocampus in regulating HPA function, any direct effects of GCs on the hippocampus will indirectly alter PVN function. See text for further details. Serotonin, 5-HT; norepinephrine, NE.
In conclusion, there is no doubt that prenatal GC treatment affords great benefit to the preterm infant. However, animal studies, now performed in many species, indicate that there may be some long-term physiologic consequences of early exposure to excess GC, and that these may be sex specific. Furthermore, the effects may not become apparent until later life. Given the dynamics of corticosteroid receptor systems in late gestation, it is likely that there are critical windows of development when specific regions of the brain are more sensitive to the influence of exogenous GC. Once such windows have been identified, it will be possible to target prenatal treatments so as to maximize benefits and reduce risks of long-term effects. Notwithstanding, the data reviewed indicate that some caution should be exercised in the use of multiple-course GC therapy during pregnancy.
Abbreviations
- AVP:
-
vasopressin
- CRH:
-
corticotropin-releasing hormone
- GC:
-
glucocorticoid
- GR:
-
glucocorticoid receptor
- HPA:
-
hypothalamo-pituitary-adrenal
- MR:
-
mineralocorticoid receptor
- PVN:
-
paraventricular nucleus
- 11β-HSD:
-
11β-hydroxysteroid dehydrogenase
References
Gould E 1994 The effects of adrenal steroids and excitatory input on neuronal birth and survival. Ann NY Acad Sci 743: 73–94
Gould E, Woolley CS, McEwen BS 1991 Adrenal steroids regulate postnatal development of the rat dentate gyrus: I. J Comp Neurol 313: 479–485
Oppenheimer JH, Schwartz HL 1997 Molecular basis of thyroid hormone-dependent brain development. Endocr Rev 18: 462–475
Bohn MC 1984 Glucocorticoid induced teratologies of the nervous system. In: Yanai J (ed) Neurobehavioral Teratology. Elsevier Science Publishers, New York, pp 365–387
Liggins GC, Howie RN 1972 A controlled trial of antepartum glucocorticoid treatment for the prevention of the respiratory distress syndrome in premature infants. Pediatrics 50: 515–523
NIH Consensus development conference. 1995 Effect of corticosteroids for fetal maturation and perinatal outcomes. Am J Obstet Gynecol 173: 253–344
Dodic M, Peers A, Coghlan JP, Wintour M 1999 Can excess glucocorticoid, in utero, predispose to cardiovascular and metabolic disease in middle age?. Trends Endocrinol Metab 10: 86–91
Takahashi LK 1998 Prenatal stress: consequences of glucocorticoids on hippocampal development and function. Int J Dev Neurosci 16: 199–207
Weinstock M 1997 Does prenatal stress impair coping and regulation of hypothalamic-pituitary-adrenal axis. Neurosci Biobehav Rev 21: 1–10
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M 1998 Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: 269–301
Beggs JM, Brown TH, Byrne JH, Crow T, LeDoux JE, LeBar K, Thompson RF 1999 Learning and memory: basic mechanisms. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR (eds) Fundamental Neuroscience. Academic Press, San Diego, pp 1411–1454
Jacobson L, Sapolsky R 1991 The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev 12: 118–134
Levine S 1957 Maternal and environmental influences on the adrenocortical response to stress in weanling rats. Science 156: 258–260
Herman JP, Prewitt CM, Cullinan WE 1996 Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stress axis. Crit Rev Neurobiol 10: 371–394
Munck A, Guyre PM, Holbrook NJ 1984 Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev 5: 25–44
Lupien SJ, McEwen BS 1997 The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Brain Res Rev 24: 1–27
Dobbing J, Sands J 1973 Quantitative growth and development of human brain. Arch Dis Child 48: 757–767
Dobbing J, Sands J 1979 Comparative aspects of the brain growth spurt. Early Hum Dev 3: 79–83
Matthews SG, Challis JRG 1996 Regulation of the hypothalamo-pituitary-adrenocortical axis in fetal sheep. Trends Endocrinol Metab 7: 239–246
Matthews SG 1998 Dynamic changes in glucocorticoid and mineralocorticoid receptor mRNA in the developing guinea pig brain. Dev Brain Res 107: 123–132
Sapolsky RM, Meaney MJ 1986 Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res Rev 396: 64–76
Claman HN 1972 Corticosteroids and lymphoid cells. N Engl J Med 287: 388–397
Dalle M, Delost P 1979 Foetal-maternal production and transfer of cortisol during the last days of gestation in the guinea-pig. J Endocrinol 82: 43–51
Edwards CRW, Benediktsson R, Lindsay RS, Seckl JR 1996 11β-Hydroxysteroid dehydrogenases: key enzymes in determining tissue-specific glucocorticoid effects. Steroids 61: 263–269
Seckl JR 1997 Glucocorticoids, feto-placental 11β-hydroxysteroid dehydrogenase type 2, and the early life origins of adult disease. Steroids 62: 89–94
Benediktsson R, Calder AA, Edwards CRW, Seckl JR 1997 Placental 11β-hydroxysteroid dehydrogenase: a key regulator of fetal glucocorticoid exposure. Clin Endocrinol Oxf 46: 161–166
Sampath-Kumar R, Matthews SG, Yang K 1998 11β-hydroxysteroid dehydrogenase type 2 is the predominant isozyme in the guinea pig placenta: decreases in messenger ribonucleic acid and activity at term. Biol Reprod 59: 1378–1384
Stewart PM, Rogerson FM, Mason JI 1995 Type 2: 11β-hydroxysteroid dehydrogenase messenger ribonucleic acid and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal adrenal steroidogenesis. J Clin Endocrinol Metab 80: 885–890
Antoni FA 1986 Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin releasing hormone. Endocr Rev 7: 351–378
Whitnall MH 1993 Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog Neurobiol 40: 573–629
Plotsky PM 1991 Pathways to the secretion of adrenocorticotropin: a view from the portal. J Neuroendocrinol 3: 1–9
Miller WL, Tyrrell JB 1995 The adrenal cortex. In: Felig P, Baxter JD, Frohman LA (eds) Endocrinology and Metabolism. McGraw Hill, New York, pp 555–711
Seeman TE, Robbins RJ 1994 Aging and hypothalamic-pituitary-adrenal response to challenge in humans. Endocr Rev 15: 233–260
Plotsky PM, Thrivikraman KV, Meaney MJ 1993 Central and feedback regulation of hypothalamic corticotropin-releasing factor secretion. Ciba Found Symp 172: 59–75
Dallman MF, Akana SF, Levin N, Walker C-D, Bradbury MJ, Suemaru S, Scribner KS 1994 Corticosteroids and the control of function in the hypothalamo-pituitary-adrenal (HPA) axis. Ann NY Acad Sci 746: 22–32
Krozowski ZS, Funder JW 1983 Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA 80: 6056–6060
Levitt NS, Lindsay RS, Holmes MC, Seckl JR 1996 Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology 64: 412–418
Muneoka K, Mikuni M, Ogawa T, Kitera K, Kamei K, Takigawa M, Takahashi K 1997 Prenatal dexamethasone exposure alters brain monoamine metabolism and adrenocortical response in rat offspring. Am J Physiol 273: R1669–R1675
Bakker JM, Schmidt ED, Kroes H, Kavelaars A, Heijnen CJ, Tilders FJH, Van Rees EP 1995 Effects of short-term dexamethasone treatment during pregnancy on the development of the immune system and the hypothalamo-pituitary adrenal axis in the rat. J Neuroimmunol 63: 183–191
Dean F, Yu C, Matthews SG 1999 Maternal glucocorticoid treatment in late gestation programs hypothalamo-pituitary-adrenal function in guinea pig offspring. Endocrine Society, San Diego, Abstract P. 2: 397
Dean F, Matthews SG 1999 Maternal dexamethasone treatment in late gestation alters glucocorticoid and mineralocorticoid receptor mRNA in the fetal guinea pig brain. Brain Res 846: 253–259
Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, Holden J 1994 Neurotoxicity of glucocorticoids in the primate brain. Horm Behav 28: 336–348
Brindley DN, Rolland Y 1989 Possible connections between stress, diabetes, obesity, hypertension and altered lipoprotein metabolism that may result in atherosclerosis. Clin Sci 77: 453–461
Munck A, Náray-Fejes-Tóth A 1994 Glucocorticoids and stress: permissive and suppressive actions. Ann NY Acad Sci 746: 115–120
Ader R, Grota LJ 1973 Adrenocortical mediation of the effects of early life experiences. Prog Brain Res 39: 395–406
Diaz R, Fuxe K, Ögren SO 1997 Prenatal corticosterone treatment induces long-term changes in spontaneous and apomorphine-mediated motor activity in male and female rats. Neuroscience 81: 129–140
Holson RR, Gough B, Sullivan P, Badger T, Sheehan DM 1995 Prenatal dexamethasone or stress but not ACTH or corticosterone alter sexual behavior in male rats. Neurotoxicol Teratol 17: 393–401
Gandelman R, Rosenthal C 1981 Deleterious effects of prenatal prednisolone exposure upon morphological and behavioral development of mice. Teratology 24: 293–301
Rayburn WF, Christensen HD, Gonzalez CL 1997 A placebo-controlled comparison between betamethasone and dexamethasone for fetal maturation: differences in neurobehavioral development of mice offspring. Am J Obstet Gynecol 176: 842–850
Rayburn WF, Christensen CL, Gonzalaz LA, Rayburn LA, Stewart JD 1998 Effect of in utero exposure to betamethasone on motivation/anxiety testing in mice offspring. Neurotoxicol Teratol 20: 475–481
Salokorpi T, Sajaniemi N, Hällback H, Kari A, Rita H, Von Wendt L 1997 Randomized study of the effect of antenatal dexamethasone on growth and development of premature children at the corrected age of 2 years. Acta Paediatr 86: 294–298
MacArthur BA, Howie RN, Dezoete JA, Elkins J 1981 Cognitive and psychosocial development of 4-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics 68: 638–643
MacArthur BA, Howie RN, Dezoete JA, Elkins J 1982 School progress and cognitive development of 6-year-old children whose mothers were treated antenatally with betamethasone. Pediatrics 70: 99–105
Smolders-de Hass H, Neuvel J, Schmand B, Treffers PE, Koppe JG, Hoeks J 1990 Physical development and medical history of children who were treated antenatally with corticosteroids to prevent respiratory distress syndrome: a 10- to 12-year follow-up. Pediatrics 86: 65–70
Dammann O, Walther H, Allers B, Schröder M, Drescher J, Lutz D, Veelken N, Schulte FJ 1996 Development of a regional cohort of very-low-birthweight children at six years: cognitive abilities are associated with neurological disability and social background. Dev Med Child Neurol 38: 97–108
Goldenberg RL, DuBard MB, Cliver SP, Nelson KG, Blankson K, Ramey SL, Herman A 1996 Pregnancy outcome and intelligence at age five years. Am J Obstet Gynecol 175: 1511–1515
McCarton CM, Wallace IF, Divon M, Vaughan HG Jr 1996 Cognitive and neurologic development of the premature, small for gestational age infant through age 6: comparison by birth weight and gestational age. Pediatrics 98: 1167–1178
Breslau N, Chilcoat H, DelDotto J, Andreski P, Brown G 1996 Low birth weight and neurocognitive status at six years of age. Biol Psychiatry 40: 389–397
Trautman PD, Meyer-Bahlburg HFL, Postelnek J, New MI 1995 Effects of early prenatal dexamethasone on the cognitive and behavioural development of young children: results of a pilot study. Psychoneuroendocrinology 20: 439–449
French NP, Hagan R, Evans SF, Godfrey M, Newnham JP 1999 Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol 180: 114–121
Bohn MC, Dean D, Hussain S, Giuliano R 1994 Development of mRNAs for glucocorticoid and mineralocorticoid receptors in rat hippocampus. Dev Brain Res 77: 157–162
Rosenfeld P, van Eekelen JAM, Levine S, De Kloet ER 1993 Ontogeny of corticosteroid receptors in the brain. Cell Mol Neurobiol 13: 295–319
Meaney MJ, Sapolsky RM, McEwen BS 1985 The development of the glucocorticoid receptor system in the rat limbic brain: II. Dev Brain Res 18: 165–168
van Eekelen JAM, Bohn MC, De Kloet ER 1991 Postnatal ontogeny of mineralocorticoid and glucocorticoid receptor gene expression in regions of the rat tel- and diencephalon. Dev Brain Res 61: 33–43
Cintra A, Solfrini V, Bunnemann B, Okret S, Bortolotti F, Gustafsson J-Å, Fuxe K 1993 Prenatal development of glucocorticoid receptor gene expression and immunoreactivity in the rat brain and pituitary gland: a combined in situ hybridization and immunocytochemical analysis. Neuroendocrinology 57: 1133–1147
Kitraki E, Alexis MN, Papalopoulou M, Stylianopoulou F 1996 Glucocorticoid receptor gene expression in the embryonic rat brain. Neuroendocrinology 63: 305–317
Diaz R, Brown RW, Seckl JR 1998 Distinct ontogeny of glucocorticoid and mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase types I and II mRNAs in the fetal rat brain suggest a complex control of glucocorticoid actions. J Neurosci 18: 2570–2580
Jones CT, Roebuck MM 1980 The development of the pituitary-adrenal axis in the guinea pig. Acta Endocrinol 94: 107–116
Andrews MH, Dean F, Matthews SG 1998 Hypothalamic glucocorticoid receptor and heat shock protein 70 expression during development in sheep: implications for negative feedback. Endocrine Society New Orleans 1: 401
Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, Perlmann T, Lehmann JM 1998 An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92: 73–82
Plotsky PM, Cunningham ETJ, Widmaier EP 1989 Catecholaminergic modulation of corticotropin-releasing factor and adrenocorticotropin secretion. Endocr Rev 10: 437–458
Cunningham ETJ, Sawchenko PE 1988 Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. J Comp Neurol 274: 60–76
Cunningham ETJ, Bohn MC, Sawchenko PE 1990 Organization of adrenergic inputs to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Comp Neurol 292: 651–667
Nakane H, Shimizu N, Hori T 1994 Stress-induced norepinephrine release in the rat prefrontal cortex measured by microdialysis. Am J Physiol 257: R1559–R1567
Nisenbaum LK, Abercrombie ED 1993 Presynaptic alterations associated with enhancement of evoked release and synthesis of norepinephrine in hippocampus of chronically cold-stressed rats. Brain Res 608: 280–287
Yau JLW, Seckl JR 1992 Central 6-hydroxydopamine lesions decrease mineralocorticoid, but not glucocorticoid receptor gene expression in the rat hippocampus. Neurosci Lett 142: 159–162
Slotkin TA, Lappi SE, McCook EC, Tayyeb MI, Eylers JP, Seidler FJ 1992 Glucocorticoids and the development of neuronal function: effects of prenatal dexamethasone exposure on central noradrenergic activity. Biol Neonate 61: 326–336
Slotkin TA, Lappi SE, Tayyeb MI, Seidler FJ 1991 Dose-dependent glucocorticoid effects on noradrenergic synaptogenesis in rat brain: ontogeny of [3H]desmethylimipramine binding sites after fetal exposure to dexamethasone. Res Commun Chem Pathol Pharmacol 73: 3–19
Barbazanges A, Piazza PV, Le Moal M, Maccari S 1996 Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci 16: 3943–3949
Sawchenko PE, Swanson LW, Steinbush HW, Verhofstad AA 1983 The distribution and cells of origin of serotonergic inputs to the paraventricular and supraoptic nuclei of the rat. Brain Res 277: 355–360
Saphier D, Feldman S 1989 Paraventricular nucleus neuronal responses following electrical stimulation of the midbrain dorsal raphe: evidence for cotransmission. Exp Brain Res 78: 407–414
Gibbs DM, Vale WW 1983 Effect of the serotonin re-uptake inhibitor fluoxetine on corticotropin releasing factor and vasopressin secretion into the hypophyseal portal blood. Brain Res 280: 176–179
Calogero AE, Bagdy G, Moncada ML, D'Agata R 1993 Effect of selective serotonin agonists on basal, corticotrophin-releasing hormone- and vasopressin-induced ACTH release in vitro from rat pituitary cells. J Endocrinol 136: 381–387
Kageyama K, Tozawa F, Horiba N, Watanobe H, Suda T 1998 Serotonin stimulates corticotropin-releasing factor gene expression in the hypothalamic paraventricular nucleus of conscious rats. Neurosci Lett 243: 17–20
Hayashi A, Nagaoka M, Yamada K, Ichitani Y, Miake Y, Okado N 1998 Maternal stress induces synaptic loss and developmental disabilities of offspring. Int J Dev Neurosci 16: 209–216
Slotkin TA, Barnes GA, McCook EC, Seidler FJ 1996 Programming of brainstem serotonin transporter development by prenatal glucocorticoids. Dev Brain Res 93: 155–161
Semont A, Fache MP, Ouafik L, Hery M, Faudon M, Hery F 1999 Effect of serotonin inhibition on glucocorticoid and mineralocorticoid expression in various brain structures. Neuroendocrinology 69: 121–128
Bamberger CM, Schulte HM, Chrousos GP 1996 Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 17: 245–261
Ajilore OA, Sapolsky RM 1999 In vivo characterization of 11β-hydroxysteroid dehydrogenase in rat hippocampus using glucocorticoid neuroendangerment as an endpoint. Neuroendocrinology 69: 138–144
Seckl JR 1997 11β-Hydroxysteroid dehydrogenase in the brain: a novel regulator of glucocorticoid action?. Front Neuroendocrinol 18: 49–99
Bodine PV, Litwack G 1990 The glucocorticoid receptor and its endogenous regulators. Receptor 1: 83–119
Orti E, Bodwell JE, Munck A 1992 Phosphorylation of steroid hormone receptors. Endocr Rev 13: 105–128
Sanchez ER, Hu J-L, Zhong S, Shen P, Greene MJ, Housley PR 1994 Potentiation of glucocorticoid receptor-mediated gene expression by heat and chemical shock. Mol Endocrinol 8: 408–421
Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato ACB 1994 A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J 13: 4087–4095
Webster JC, Cidlowski JA 1994 Downregulation of the glucocorticoid receptor: a mechanism for physiological adaptation to hormones. Ann NY Acad Sci 746: 216–220
Silva CM, Powell-Oliver F, Jewell CM, Sar M, Allgood VE, Cidlowski JA 1994 Regulation of the human glucocorticoid receptor by long-term and chronic treatment with glucocorticoid. Steroids 59: 436–442
Kalinyak JE, Griffin CA, Hamilton RW, Bradshaw JG, Perlman AJ, Hoffman AR 1989 Developmental and hormonal regulation of glucocorticoid receptor messenger RNA in the rat. J Clin Invest 84: 1843–1848
Meaney MJ, Sapolsky RM, McEwen BS 1985 The development of the glucocorticoid receptor system in the rat limbic brain: I. Dev Brain Res 18: 159–164
Mitchell JB, Rowe W, Boksa P, Meaney MJ 1990 Serotonin regulates type II corticosteroid receptor binding in hippocampal cell cultures. J Neurosci 10: 1745–1752
Anisman H, Zaharia MD, Meaney MJ, Merali Z 1998 Do early-life events permanently alter behavioral and hormonal responses to stressors?. Int J Dev Neurosci 16: 149–164
Meaney MJ, Diorio J, Francis D, Larocque S, O'Donnell D, Smythe JW, Sharma S, Tannenbáum B 1994 Environmental regulation of the development of glucocorticoid receptor systems in the rat forebrain: the role of serotonin. Ann NY Acad Sci 746: 260–274
Mitchell JB, Iny LJ, Meaney MJ 1990 The role of serotonin in the development and environmental regulation of type II corticosteroid receptor binding in rat hippocampus. Dev Brain Res 55: 231–235
Meaney MJ, Aitken DH, Sapolsky RM 1987 Thyroid hormones influence the development of hippocampal glucocorticoid receptors in the rat: a mechanism for the effects of postnatal handling on the development of the adrenocortical stress response. Neuroendocrinology 45: 278–283
Sapolsky RM 1999 Why stress is bad for your brain. Science 273: 749–750
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW 1996 Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 93: 3908–3913
Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, Delaney RC, McCarthy G, Charney DS, Innis RB 1995 MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152: 973–981
Stein MB, Koverola C, Hanna C, Torchia MG, McClarty B 1997 Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 27: 951–959
Uno H, Lohmiller L, Thieme C, Kemnitz JW, Engle MJ, Roecker EB, Farrell PM 1990 Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques: I. Dev Brain Res 53: 157–167
De Kloet ER, Rosenfeld P, Van Eikelen JA, Sutanto W, Levine S 1988 Stress, glucocorticoids and development. Prog Brain Res 73: 101–120
Sapolsky RM 1996 Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress 1: 1–19
Horner HC, Packan DR, Sapolsky RM 1990 Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology 52: 57–64
Lawrence MS, Sapolsky RM 1994 Glucocorticoids accelerate ATP loss following metabolic insults in cultured hippocampal neurons. Brain Res 646: 303–306
Dunlop SA, Archer MA, Quinlivan JA, Beazley LD, Newnham JP 1997 Repeated prenatal corticosteroids delay myelination in the ovine central nervous system. J Matern Fetal Med 6: 309–313
Carlos RQ, Seidler FJ, Slotkin TA 1992 Fetal dexamethasone exposure alters macromolecular characteristics of rat brain development: a critical period for regionally selective alterations?. Teratology 46: 45–59
Carlos RQ, Seidler FJ, Lappi SE, Slotkin TA 1991 Fetal dexamethasone exposure affects basal ornithine decarboxylase activity in developing rat brain regions and alters acute responses to hypoxia and maternal separation. Biol Neonate 59: 69–77
Rodriguez JJ, Montaron MF, Petry KG, Aurousseau C, Marinelli M, Premier S, Rougon G, Le Moal M, Abrous DN 1998 Complex regulation of the expression of the polysialylated form of the neuronal cell adhesion molecule by glucocorticoids in the rat hippocampus. Eur J Neurosci 10: 2994–3006
Rose SP 1995 Cell adhesion molecules, glucocorticoids and long-term memory formation. Trends Neurosci 18: 502–506
Slotkin TA, Zhang J, McCook EC, Seidler FJ 1998 Glucocorticoid administration alters nuclear transcription factors in fetal rat brain: implications for the use of antenatal steroids. Brain Res Dev Brain Res 111: 11–24
Slotkin TA, Lau C, Lappi FJ, Seidler FJ 1996 Can intracellular signaling pathways predict developmental abnormalities? Sensitivity of the adenyl cyclase/c-fos proto-oncogene cascade to β-adrenergic agonists and glucocorticoids in the fetal rat. Biomarkers 1: 115–122
Lindsay RM, Wiegand SJ, Altar CA, DiStefano PS 1994 Neurotropic factors: from molecule to man. Trends Neurosci 17: 182–190
Collazo D, Takahashi H, McKay RD 1992 Cellular targets and trophic functions of neurotrophin-3 in the developing rat hippocampus. Neuron 9: 643–656
Smith MA, Makino S, Kvetnansky R, Post RM 1995 Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15: 1768–1777
Smith MA, Makino S, Kim SY, Kvetnansky R 1995 Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology 136: 3743–3750
Smith MA 1996 Hippocampal vulnerability to stress and aging: possible role of neurotrophic factors. Behav Brain Res 78: 25–36
Chao HM, McEwen BS 1994 Glucocorticoids and the expression of mRNAs for neurotrophins, their receptors and GAP-43 in the rat hippocampus. Brain Res Mol Brain Res 26: 271–276
Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD 1990 NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5: 501–509
Seckl JR, Olsson T 1995 Glucocorticoid hypersecretion and the age-impaired hippocampus: cause or effect. J Endocrinol 145: 201–211
Marinoni E, Korebrits C, Di Iorio R, Cosmi EV, Challis JR 1998 Effect of betamethasone in vivo on placental corticotropin-releasing hormone in human pregnancy. Am J Obstet Gynecol 178: 770–778
Marinoni E, Di Iorio R, Letizia C, Villaccio B, Scucchi L, Cosmi EV 1998 Immunoreactive adrenomedullin in human fetoplacental tissues. Am J Obstet Gynecol 179: 784–787
Clifton VL, Owens PC, Robinson PJ, Smith R 1995 Identification and characterization of a corticotrophin-releasing hormone receptor in human placenta. Eur J Endocrinol 133: 591–597
Clifton VL, Read MA, Leitch IM, Boura ALA, Robinson PJ, Smith R 1994 Corticotropin-releasing hormone-induced vasodilatation in the human fetal placental circulation. J Clin Endocrinol Metab 79: 666–669
Author information
Authors and Affiliations
Additional information
Supported by the Medical Research Council of Canada (MT-1469) and the Natural Sciences and Engineering Council of Canada.
Rights and permissions
About this article
Cite this article
Matthews, S. Antenatal Glucocorticoids and Programming of the Developing CNS. Pediatr Res 47, 291–300 (2000). https://doi.org/10.1203/00006450-200003000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200003000-00003
This article is cited by
-
Improving behavioral deficits induced by perinatal ethanol and stress exposure in adolescent male rat progeny via maternal melatonin treatment
Psychopharmacology (2024)
-
Antenatal Dexamethasone Treatment Induces Sex-dependent Upregulation of NTPDase1/CD39 and Ecto-5ʹ-nucleotidase/CD73 in the Rat Fetal Brain
Cellular and Molecular Neurobiology (2022)
-
Long-term neuropathological and/or neurobehavioral effects of antenatal corticosteroid therapy in animal models: a systematic review
Pediatric Research (2020)
-
Interaction between Prenatal Maternal Stress and Autonomic Arousal in Predicting Conduct Problems and Psychopathic Traits in Children
Journal of Psychopathology and Behavioral Assessment (2017)
-
Early life programming of pain: focus on neuroimmune to endocrine communication
Journal of Translational Medicine (2016)
