
Abstract
Placental abnormalities reflect antenatal disease processes that may interact with other perinatal risk factors to affect long-term outcome. We performed a nested case control analysis of placental and clinical risk factors associated with neurologic impairment (NI) at 20-mo corrected age (60 cases and 59 controls) using data collected in a prospective study of very low birth weight (less than 1500 g) infants born between 1983 and 1991. In a preliminary analysis we explored the relationship between clinical infection and histologic chorioamnionitis (CA). Only histologic CA with a fetal vascular response correlated with either clinical CA or early onset neonatal sepsis. We then assessed the relative contribution of the nine risk factors (four placental and five clinical) associated with NI at the univariate level by multiple logistic regression. Three risk factors were independent predictors of NI: severe cranial ultrasound abnormalities (odds ratio 13.6, 95% confidence intervals 4.5–66.7), multiple placental lesions (odds ratio 13.2, 95% confidence intervals 1.3–137.0), and oxygen dependence at 36 wk (odds ratio 4.2, 95% confidence intervals 1.2–14.6). Finally, a series of logistic regressions was conducted with the dependent variable changing as we-moved back along the causal chain to explore the relationships between risk factors operating at different stages. This analysis suggested that antenatal variables that were not independent predictors of NI by multiple logistic regression exerted their effects through the following intermediate pathways: fetal grade 3 histologic CA via chorionic vessel thrombi, clinical CA via grade 3 villous edema, and grade 3 villous edema via severe cranial ultrasound abnormalities.
Similar content being viewed by others

Main
The increased survival of very low birth weight (VLBW) infants (<1.5 kg) has resulted in a marked change in the epidemiology of cerebral palsy and related forms of neurologic impairment (NI). Surviving VLBW infants currently account for 25–50% of all cases (1, 2). Several recent studies have addressed perinatal risk factors for NI in these infants. Parenchymal brain lesions such as severe intraventricular hemorrhage with periventricular infarction and periventricular leukomalacia detected by neonatal imaging studies are the strongest correlates (3–5). Other important risk factors for NI include amniotic fluid infections, hypocarbia, hypothyroxinemia, prolonged rupture of membranes, and the development of chronic lung disease (6–11).
In a case control study of VLBW infants delivered and followed at Rainbow Babies and Children's Hospital over the period 1983–1991 we identified clinical risk factors associated with NI at 20-mo corrected gestational age (12). We subsequently described the placental pathology of these infants (13). Placental lesions may act directly to cause brain injury or indirectly by increasing the incidence of neonatal complications. To distinguish between these alternatives we analyzed the interrelationships between placental and clinical risk factors and assessed their relative and temporal contributions to NI. In view of recent interest in the role of amniotic fluid infections in brain injury, we also explored the relationship between histologic CA and clinical CA, their associations with neonatal sepsis, and the mechanisms by which each contributes to NI (14, 15).
MATERIALS AND METHODS
Study population.
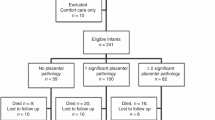
This was a nested case control analysis of placental and clinical risk factors associated with NI at 20-mo corrected age in singleton infants without major congenital anomalies using data collected in a prospective study of VLBW (<1500 g) infants born between 1983 and 1991 at our institution and followed in a high-risk follow-up program at Rainbow Babies and Children's Hospital. In a previous study, 72 with major NI at 20-mo corrected age (50 infants with strictly defined cerebral palsy plus 22 infants with other neurologic abnormalities that overlap with the diagnosis of cerebral palsy—hypertonia, defined as increased muscle tone, usually in the lower extremities with normal truncal tone; hypotonia, defined as decreased muscle tone involving the trunk and/or extremities; and shunt dependent hydrocephalus without abnormal neurologic signs—were compared with 72 controls with a normal neurologic examination at 20-mo corrected age selected among infants born in the same year and matched for weight (±250 g), gestational age (+ 2 wk), and race (12).
The population evaluated in this study was the subset of cases and controls from the original sample in whom placentas were submitted to pathology (13). Placental reports and slides were available in 119 of the 144 original study patients [strict cerebral palsy—42/50 (84%), strict cerebral palsy controls—43/50 (86%), other neurologic abnormalities—18/22 (82%), and other neurologic abnormalities controls—16/22 (73%)]. Comparisons among the original matching variables in patients with placentas available for study were as follows: African-American mothers—57% for cases versus 59% for controls; mean gestational age—27.3 ± 2.3 wk for cases and 27.5 ± 2.2 wk for controls; and mean birth weight—978 ± 228 g for cases versus 1012 ± 226 g for controls. None of the above differences were statistically significant.
Clinical findings.
The significance of clinical determinants of NI from our previous study was recalculated for the subsample with placental pathology. Of the determinants identified as associated with NI in the previous study, only hypothyroxinemia had a p value >0.10 in the subsample. The definitions of the clinical findings evaluated in this report have been previously described and are briefly summarized below (12). Clinical CA was defined as premature rupture of membranes with a temperature of either >37.8°C on two occasions, at least 1 h apart, or >38.3°C on one occasion with no other known sources for the fever and one of the following: maternal tachycardia (≥100 beats per min), fetal tachycardia (≥160 beats per min), maternal leukocytosis (>11 000/mm white blood cells), or foul-smelling amniotic fluid. Chronic lung disease was defined by oxygen dependence at 36-wk postconceptional age. Sepsis was defined by at least one positive blood culture accompanied by clinical signs suggestive of infection. Sepsis was subdivided into early sepsis (<7 d after birth) and late sepsis (≥7 d after birth) (16). Hyperbilirubinemia was defined as a maximum total bilirubin level >10 mg/dL. Severe cranial ultrasound abnormality was defined as the diagnosis of one or more of the following at any time during admission: grade 3–4 periventricular hemorrhage, parenchymal infarction, or periventricular leukomalacia, or the presence of ventricular dilatation at discharge.
Placental pathology.
All placentas were examined and processed using a standard protocol (17). Details of the examination and classification scheme have previously been described (13) and conform to published consensus criteria (18). Chorionic vessel thrombi were defined by laminated fibrin adherent to the wall of chorionic vessels. Severe (grade 3) villous edema was defined as large vacuoles in the stroma affecting >19% of villi. Histologic CA was separated into maternal and fetal inflammatory components and graded as follows: Maternal grade, based on the worst area of the chorionic plate: 1—rare neutrophils [<10/high-powered field (HPF) ], 2—intermediate neutrophils (11–30/HPF), and 3—abundant neutrophils (>30/HPF). Fetal grade, based on the most severely affected chorionic plate vessel: 1—rare neutrophils, 2—intermediate neutrophils, and 3—abundant neutrophils. The category of multiple placental lesions was defined by the presence of two or more of the following lesions: retroplacental hematoma with indentation, multiple villous infarcts, diffuse microinfarction, increased nucleated red blood cells, and grade 2–3 increased basal perivillous fibrin.
Statistical analysis.
χ2 and Fisher exact tests were used for nominal data. To test the independent effects of placental and other perinatal risk factors, unconditional logistic regression was conducted. Due to limited sample size we included only variables with univariate associations at the p < 0.10 level. Variables were separated into three groups: prenatal, neonatal, and sonographic. To examine the effects of these three groups of findings, each group of variables was added by block entry as a separate step in the logistic regression analysis. Finally, we used methodology drawn from the principles of path analysis to temporally examine the relationships between antecedents of NI (19). A series of logistic regression analyses was conducted, with the dependent variable changing as we moved back along the causal chain, including all variables that could be considered as antecedent to that dependent variable. The groupings for this analysis were findings indicating antenatal infection, placental lesions, and neonatal variables. The purpose of these analyses was not to calculate indirect or total effects as done in linear-based path analysis, but rather to provide a conceptual framework for exploring interrelationships.
RESULTS
The frequencies of all placental and risk factors significant at the p > 0.10 level in VLBW infants with and without NI at 20-mo corrected age is shown in Table 1 with their associated odds ratios and 95% confidence intervals. Placental abnormalities that increased in infants with NI were chorionic vessel thrombi, severe (grade 3) villous edema, severe fetal (grade 3) histologic CA, and multiple placental lesions. Clinical findings that increased in infants with NI were clinical CA, oxygen dependence at 36-wk postconceptional age, sepsis, hyperbilirubinemia (>10 mg/dL), and severe cranial ultrasound abnormalities.
Interrelationships among risk factors indicative of perinatal infection (histologic CA, clinical CA, and sepsis) are illustrated for descriptive purposes in Table 2. Four histologic subgroups were distinguished: 1—no histologic CA, 2—histologic CA without fetal inflammation (maternal inflammation only), 3—histologic CA with mild-to-moderate (grades 1–2) fetal inflammation, and 4—histologic CA with severe (grade 3) fetal inflammation. Groups 1 and 2 (no histologic CA and histologic CA without fetal inflammation) had equivalent low levels of concordance with clinical CA (9% and 7%, respectively). Group 3 (histologic CA with mild-to-moderate fetal inflammation) showed only slightly better concordance (15%). Only Group 4 (histologic CA with severe fetal inflammation) had a high level of concordance (77%) with clinical CA. Early sepsis (<7 d after birth) was uncommon (6% of total cases) and occurred only in infants whose placentas had histologic CA with fetal inflammation (groups 3 and 4). Late sepsis (≥7 d after birth) was unrelated to histologic CA. The relative effects of clinical CA and/or histologic grade 3 CA on the risk of NI are summarized at the bottom of Table 2. While fetal grade 3 histologic CA was associated with clinical CA as described above, more infants (54%) had one or the other finding than both. Odds ratios for NI were higher with clinical CA alone (OR 6.0) and fetal grade 3 histologic CA (OR 5.3) than they were when both were present (OR 2.1). Because of the small numbers involved and the retrospective nature of the data, no statistical inferences were drawn from this unexpected finding.
To determine independent predictors of NI, logistic regression with block entry of prenatal, postnatal, and sonographic risk factors was performed (Table 3). In the full analysis including all three groups of variables (Model 3), three risk factors—multiple placental lesions, oxygen dependence at 36-wk postconceptional age, and severe cranial ultrasound abnormalities—were found to be independent predictors of NI. After exclusion of findings obtained by direct imaging of the CNS (Model 2), grade 3 villous edema achieved significance. Chorionic vessel thrombi, while not significant, maintained adjusted odds ratios (3.5–3.8) similar to the unadjusted odds ratio (3.7) shown in Table 1 in all three models. Clinical CA, fetal grade 3 histologic CA, hyperbilirubinemia, and sepsis were not independent predictors of NI in any of the models tested.
Finally, interrelationships among risk factors operating at different stages were explored. A series of logistic regressions was conducted with the dependent variable changing as we moved back along the causal chain, considering all variables that might be considered antecedent to that dependent variable. The temporal groupings for this analysis were 1—findings indicative of antenatal infection, 2—placental lesions, and 3—neonatal variables. The rationale for the ordering of groups 1 and 2 is that, although antenatal infection could be an antecedent for some of the placental lesions in group 2, the converse is highly unlikely. Variables in groups 1 and 2, occurring before birth, were clearly antecedent to neonatal variables in Group 3. Figure 1 illustrates the groupings and the relationships between variables with their associated odds ratios and 95% confidence intervals. Only relationships significant at p < 0.45 are shown. In addition to the predictors of NI described above, four significant associations between intermediary variables were found: grade 3 villous edema with severe ultrasound abnormalities, clinical CA with grade 3 villous edema, fetal grade 3 histologic CA with chorionic vessel thrombi, and fetal grade 3 histologic CA with clinical CA. Oxygen dependence at 36-wk postconceptional age showed weak nonsignificant relationships to two other variables—grade 3 villous edema and clinical CA.

Direct and mediating effects of risk factors for NI as determined by serial logistic regressions (see “Materials and Methods”). Variables were considered in three groups: indicators of antenatal infection, placental abnormalities not directly attributable to bacterial infection, and neonatal factors. Significant interrelationships are indicated by solid lines with odds ratios and 95% confidence intervals. All nonsignificant interrelationships at the p < 0.450 level are also shown (dashed lines). Abbreviations are as follows (see “Materials and Methods” for definitions): NI, neurologic impairment; Sev U/S, severe cranial ultrasound abnormalities; O236, oxygen dependence at 36-wk postconceptional age; Gr3 Edema, grade 3 villous edema; CV Thrombi, chorionic vessel thrombi; Multiple, multiple placental abnormalities; Clin CA, clinically diagnosed chorioamnionitis; FGr3 HCA, fetal grade 3 histologic chorioamnionitis.
DISCUSSION
Identifying the causes of cerebral palsy and related forms of NI in VLBW infants is complicated by several inherent difficulties (20). First, cerebral palsy is an operational definition combining an etiologically diverse group of children with nonprogressive motor disorders of central origin. Second, delayed ascertainment, the inability to diagnose cerebral palsy until long after the time of the actual injury, is particularly problematic for the VLBW infant where the diagnosis often follows a prolonged hospitalization with multisystem disease. To overcome this second difficulty, recent studies have focused on early objective parameters of brain injury such as severe ultrasound abnormalities (21, 22). However, these are surrogate outcomes not always corresponding to the population with long-term NI. Patients with severe ultrasound abnormalities may either be neurologically normal, die, or have severe multisystem disease removing them from the group of children eventually classified with NI. In this study we used a comprehensive assessment of neurologic motor function at 20-mo corrected age to define outcome.
It is widely acknowledged that NI reflects an interplay between antenatal, intrapartum, and neonatal processes (23, 24). Antenatal processes are particularly difficult to study because they are poorly understood and often estimated by proxies such as rupture of membranes or maternal fever rather than by specific processes affecting the fetoplacental unit. Evaluation of the placenta has the potential to provide insight into these antenatal processes by demonstrating anatomic lesions associated with decreased maternal substrate delivery, altered transport function, compromise of the fetal vasculature, or unrecognized inflammatory processes. Few studies have directly measured the impact of placental lesions on neurologic outcome and much of the existing data are limited by one or more of the following problems: data collection before modern intensive care (25, 26), analysis of surrogate outcomes such as ultrasound abnormalities or neuropathologic lesions (21, 27–29), and restricted emphasis on a single placental lesion (29, 30). Very few previous studies have attempted to separate direct from indirect effects and study interactions between placental and other perinatal risk factors (21, 25).
We studied a cohort of VLBW infants derived from a single institution born over a relatively confined time period and followed up in a comprehensive program to determine long-term outcome. In two previous papers we reported individual clinical and pathologic risk factors associated with NI (12, 13). In this paper we have studied the interrelationships between clinical and placental risk factors to determine independent predictors of NI and their antecedents. Weaknesses of the present study include retrospective analysis of data, the relatively small number of cases, the low incidence of some pathologic lesions, and the 8 y time period over which our cases were collected. Three independent risk factors for NI were identified: multiple placental lesions, severe cranial ultrasound abnormalities, and chronic lung disease as estimated by oxygen dependence at 36-wk postconceptional age.
The high rate of NI observed in infants having placentas with multiple placental lesions supports the concept that risk factors may act in an additive or synergistic fashion. Lesions in this category included two indicators of severely decreased uteroplacental perfusion (multiple infarcts and diffuse microinfarction), an idiopathic pregnancy disorder (increased basal perivillous fibrin, also known as “maternal floor infarction”), an indicator of premature placental separation (retroplacental hematoma with indentation), and a measure of subacute fetal hypoxia (increased nucleated red blood cells) (18). Similar additive or synergistic interactions between different types of lesions have been reported for ultrasound abnormalities associated with NI and placental lesions associated with stillbirth (31, 32). The relative importance of interactions among specific placental lesions is likely to depend on time of onset, duration, severity, and host susceptibility and requires larger studies to resolve.
The association of severe cranial ultrasound abnormalities (periventricular white matter degeneration, periventricular hemorrhagic infarction, and/or posthemorrhagic hydrocephalus) with NI has been well documented and thoroughly discussed in previous studies (3, 4, 31, 33). In some cases the severity of the ultrasound lesion is such that it represents a proxy for the outcome. Nevertheless, some patients with cranial severe ultrasound abnormalities (18% in our study) did not have NI at 20 mo and many patients with NI at 20 mo did not have these sonographic findings (40% in our study). Further data regarding the exact nature and timing of ultrasound abnormalities and/or more sophisticated imaging techniques could potentially improve the predictive power of neuroradiologic findings.
Severe villous edema showed a strong and somewhat unexpected relationship to cranial ultrasound abnormalities in our study. Villous edema was originally described by Naeye and further characterized by Kliman (34, 35). Previous data have suggested that this lesion is associated with poor Apgar scores, acidosis, antenatal and neonatal death, and neuropsychological abnormalities (36). While the exact pathogenesis of villous edema is not clear, it appears to reflect a fetal circulatory abnormality, to have a relatively rapid onset, and to resolve slowly. The lesion has been suggested to exert its effect by compression of villous arterioles (35). An alternative view is that villous edema is a marker for a more generalized loss of fetal circulatory integrity (37).
Oxygen dependence at 36-wk postconceptual age, an indicator of chronic lung disease, has been identified as a risk factor for NI in previous studies (3, 11). While none of the antenatal variables studied were significant predictors of chronic lung disease when the latter was treated as a dependent variable, two processes showed borderline significance—clinical CA (p = 0.113) and grade 3 villous edema (p = 0.090). Several studies have suggested that chorioamnionitis may be a predisposing factor for chronic lung disease (38–40). Possible roles for cytokines, proteases, and reactive oxygen intermediates produced by leukocytes aspirated into the lungs during the course of infection have been suggested as possible mechanisms (41, 42). In view of the suggestive nature of the available evidence and the emergence of new patterns of chronic lung disease owing to smaller surviving babies and new management strategies a larger study using more recent cases would be informative.
A number of studies have identified clinical CA as a risk factor for NI (6, 7, 12, 14, 25, 43, 44). An association between clinical and histologic CA in our study was found only for the subgroup of histologic CA with severe (grade 3) fetal inflammation. While there was considerable overlap between clinical CA and fetal grade 3 histologic CA, it was the cases with one or the other, but not both, that contributed most strongly to NI. The distinction between these two risk factors was supported by different associations with other placental lesions: fetal grade 3 CA with chorionic vessel thrombi and clinical CA with villous edema. One possible explanation might be that fetal grade 3 CA selects for longstanding amniotic fluid infections with intact membranes whereas clinical CA required ruptured membranes by definition. Interestingly, both short (<1 h) and prolonged (> 24 h) duration of membrane rupture have been associated with NI in previous studies (9, 14). Misclassification of extrauterine infections, which have also been associated with NI, as clinical CA is another possible explanation (45, 46).
Our finding that fetal as opposed to maternal inflammation is the critical feature linking histologic CA with NI, clinical CA, and early neonatal sepsis is in agreement with other recent studies and emphasizes the importance of the recently described fetal inflammatory response syndrome (47, 48). This syndrome was introduced to emphasize the deleterious effects of acute phase reactants, circulating cytokines, and other procoagulant molecules released into the fetal circulation during amniotic fluid infection. Two of these deleterious effects may be chorionic vessel thrombi and villous edema, as suggested by this study.
In summary, our retrospective study has identified multiple placental lesions, severe cranial ultrasound abnormalities, and chronic lung disease as independent predictors of NI in VLBW infants. We have also delineated patterns of placental injury that interact both with one another and with neonatal processes to increase the risk of NI. Confirmation of these relationships and extension to more specific subgroups of NI requires further study.
Abbreviations
- CA:
-
chorioamnionitis
- 95% CI:
-
95% confidence intervals
- NI:
-
neurologic impairment
- OR:
-
odds ratio
- VLBW:
-
very low birth weight
References
Bhushan V, Paneth N, Kiely JL 1993 Impact of improved survival of very low birth weight infants on recent secular trends in the prevalence of cerebral palsy. Pediatrics 91: 1094–1100
Pharoah POD, Platt MJ, Cooke T 1996 The changing epidemiology of cerebral palsy. Arch Dis Child 75: F169–F173
Pinto-Martin JA, Riolo S, Cnaan A, Holzman C, Susser MW, Paneth N 1995 Cranial ultrasound prediction of disabling and nondisabling cerebral palsy at age two in a low birth weight population. Pediatrics 95: 249–254
Paneth N, Rudelli R, Kazam E, Monte W 1994 Brain Damage in the Preterm Infant. MacKeith Press, London, pp 171–185
Leviton A, Paneth N 1990 White matter damage in preterm newborns—an epidemiologic perspective. Early Hum Dev 24: 1–22
Murphy DJ, Sellers S, MacKensie IZ, Yudkin PL, Johnson AM 1995 Case-control study of antenatal and intrapartum risk factors for cerebral palsy in very preterm singleton babies. Lancet 346: 1449–1454
Yoon BH, Jun JK, Romero R, Park KH, Gomez R, Choi JH, Kim IO 1997 Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1 beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am J Obstet Gynecol 177: 19–26
Fujimoto S, Togari H, Yamaguchi N, Mizutani F, Suzuki S, Sobajima H 1994 Hypocarbia and cystic periventricular leukomalacia in premature infants. Arch Dis Child 71: F107–F110
Spinillo A, Capuzzo E, Stronati M, Ometto M, Orcesi S, Fazzi E 1995 Effect of preterm premature rupture of membranes on neurodevelopmental outcome: follow up at two years of age. Br J Obstet Gynaecol 102: 882–887
Reuss ML, Paneth N, Pinto-Martin JA, Lorenz JM, Susser M 1996 The relation of transient hypothyroxinemia in preterm infants to neurologic development at two years of age. N Engl J Med 334: 821–827
Allan WC, Vohr B, Makuch RW, Katz KH, Ment LR 1997 Antecedents of cerebral palsy in a multicenter trial of indomethacin for intraventricular hemorrhage. Arch Pediatr Adolesc Med 151: 580–585
Wilson-Costello D, Borawski E, Friedman H, Redline RW, Fanaroff AA, Hack M 1998 Perinatal correlates of cerebral palsy and other neurologic impairment among very low birthweight children. Pediatrics 102: 315–322
Redline RW, Wilson-Costello D, Borawski E, Fanaroff AA, Hack M 1998 Placental lesions associated with neurologic impairment and cerebral palsy in very low birth weight infants. Arch Pathol Lab Med 122: 1091–1098
Dammann O, Leviton A 1997 Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42: 1–8
Nelson KB, Dambrosia JM, Grether JK, Phillips TM 1998 Neonatal cytokines and coagulation factors in children with cerebral palsy. Arch Neurol 44: 665–675
Berger A, Salzer HR, Weninger M, Sageder B, Aspock C 1998 Septicaemia in an Austrian neonatal intensive care unit: a 7-year analysis. Acta Paediatr 87: 1066–1069
Driscoll SG, Langston C 1991 College of American Pathologists Conference XIX on the Examination of the Placenta: Report on the Working Group on Methods for Placental Examination. Arch Pathol Lab Med 115: 704–708
Langston C, Kaplan C, Macpherson T, Manci E, Peevy K, Clark B, Murtagh C, Cox S, Glenn G 1997 Practice guideline for examination of the placenta. Arch Pathol Lab Med 121: 449–476
Loehlin JC 1998 Latent Variable Models: An Introduction to Factor, Path, and Structural Analysis, 3rd Ed. Lawrence Erlbaum, Mahway, NJ
Badawi N, Watson L, Blair E, Slee J, Haan E, Stanley F 1998 What constitutes cerebral palsy?. Dev Med Child Neurol 40: 520–527
The Developmental Epidemiology Network Investigators. 1998 The correlation between placental pathology and intraventricular hemorrhage in the preterm infant. Pediatr Res 43: 15–19
Yoon BH, Romero R, Yang SH, Jun JK, Kim IO, Choi JH, Syn HC 1996 Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am J Obstet Gynecol 174: 1433–1440
Pharoah POD 1995 Cerebral palsy and perinatal care. Br J Obstet Gynaecol 102: 356–358
The Australian and New Zealand Perinatal Societies. 1995 The origins of cerebral palsy—a consensus statement. Med J Aust 162: 85–90
Nelson KB, Ellenberg JH 1986 Antecents of cerebral palsy. N Engl J Med 315: 81–86
Naeye RL, Peters EC, Bartholomew MS, Landis JR 1989 Origins of cerebral palsy. Am J Dis Child 143: 1154–1161
Bejar R, Wozniak P, Allard M, Benirschke K, Vaucher Y, Coen R, Berry C, Schragg P, Villegas I, Resnik R 1988 Antenatal origin of neurologic damage in newborn infants. Am J Obstet Gynecol 159: 357–363
Grafe MR 1994 The correlation of prenatal brain damage with placental pathology. J Neuropathol Exp Neurol 53: 407–415
Burke CJ, Tannenberg AE 1995 Prenatal brain damage and placental infarction—an autopsy study. Dev Med Child Neurol 37: 555–562
Sander CH, Kinnane L, Stevens NG 1985 Hemorrhagic endovasculitis of the placenta: a clinicopathologic entity associated with adverse pregnancy outcome. Compr Ther 11: 66–74
Aziz K, Vickar DB, Sauve RS, Etches PC, Pain KS, Robertson CMT 1995 Province based study of neurologic disability of children weighing 500–1249 grams at birth in relation to neonatal ultrasound findings. Pediatrics 95: 837–844
Driscoll SG 1984 Autopsy following stillbirth: a challenge neglected. In: Ryder OA, Byrd ML, (eds) One Medicine. Springer-Verlag, Berlin, pp 20–31
Fletcher JM, Landry SH, Bohan TP, Davidson KC, Brookshire BL, Lachar D, Kramer LA, Francis DJ 1997 Effects of intraventricular hemorrhage and hydrocephalus on the long-term neurobehavioral development of very-low-birthweight infants. Dev Med Child Neurol 39: 596–606
Naeye RL, Maisels J, Lorenz RP, Botti JJ 1983 The clinical significance of placental villous edema. Pediatrics 71: 588–594
Kliman HJ, Jones DC, Morotti RA 1995 The rise and fall of villous edema as a function of time from initiation of intrauterine infection. Am J Obstet Gynecol 172: 312
Naeye RL 1992 Disorders of the Placenta, Fetus, and Neonate. Diagnosis and Clinical Significance. Mosby-Year Book Inc, St. Louis, pp 174–179
Redline RW 1995 Placental pathology: a neglected link between basic disease mechanisms and untoward pregnancy outcome. Curr Opin Obstet Gynecol 7: 10–15
Watterberg KL, Demers L, Scott SM, Murphy S 1996 Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 97: 210–215
Matsuda T, Nakajima T, Hattori S, Hanatani K, Fukazawa Y, Kobayashi K, Fujimoto S 1997 Necrotizing funisitis: clinical significance and association with chronic lung diseae in premature infants. Am J Obstet Gynecol 177: 1402–1407
Yoon BH, Romero R, Jun JK, Park KH, Park JD, Ghezzi F, Kim BI 1997 Amniotic fluid cytokines (interleukin-6, tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8) and the risk for the development of bronchopulmonary dysplasia. Am J Obstet Gynecol 177: 825–830
Speer CP, Groneck P 1998 Oxygen radicals, cytokines, adhesion molecules and lung injury in neonates. Semin Perinatol 3: 219–228
Jobe AH, Ikegami M 1998 Mechanisms initiating lung injury in the preterm. Early Hum Dev 53: 81–94
Grether JK, Nelson KB, Emery ES, Cummings SK 1996 Prenatal and perinatal factors and cerebral palsy in very low birth weight infants. J Pediatr 128: 407–414
O'Shea TM, Klinepeter KL, Meis PJ, Dillard RG 1998 Intrauterine infection and the risk of cerebral palsy in very-low birthweight infants. Paediatr Perinat Epidemiol 12: 72–83
Grether JK, Nelson KB 1997 Maternal infection and cerebral palsy in infants of normal birth weight. JAMA 278: 207–211
Mays J, Verma U, Klein, Tejani N 1995 Acute appendicitis in pregnancy and the occurrence of major intraventricular hemorrhage and periventricular leukomalacia. Obstet Gynecol 86: 650–652
Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM 1998 The fetal inflammatory response syndrome. Am J Obstet Gynecol 179: 194–202
Van Hoeven KH, Anyaegbunam A, Hochster H, Whitty JE, Distant J, Crawford C, Factor SM 1996 Clinical significance of increasing histologic severity of acute inflammation in the fetal membranes and umbilical cord. Pediatr Pathol Lab Med 16: 731–744
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Redline, R., Wilson-Costello, D., Borawski, E. et al. The Relationship Between Placental and Other Perinatal Risk Factors for Neurologic Impairment in Very Low Birth Weight Children. Pediatr Res 47, 721–726 (2000). https://doi.org/10.1203/00006450-200006000-00007
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200006000-00007
This article is cited by
-
Placental pathology and intraventricular hemorrhage in preterm and small for gestational age infants
Journal of Perinatology (2021)
-
Prophylactic inhibition of NF-κB expression in microglia leads to attenuation of hypoxic ischemic injury of the immature brain
Journal of Neuroinflammation (2020)
-
Term and preterm (<34 and <37 weeks gestation) placental pathologies associated with fetal growth restriction
Archives of Gynecology and Obstetrics (2010)
-
Neonatal morbidity and placental pathology
The Indian Journal of Pediatrics (2006)
-
The Histologic Fetoplacental Inflammatory Response in Fatal Perinatal Group B-Streptococcus Infection
Journal of Perinatology (2004)
