
Abstract
After transient cerebral ischemia in fetal sheep, delayed disruptions in cerebral energetics are represented by a delayed increase in cortical impedance, a progressive decrease in the concentration of oxidized cytochrome oxidase as measured by near-infrared spectroscopy, and cortical seizures. Because the production of nitric oxide (NO), a potent mediator of neuronal death, is increased during this phase, the present study investigated whether inhibition of NO synthesis could ameliorate the delayed disruption in cerebral energetics. Eleven late gestation fetal sheep were subjected to 30 min of transient cerebral ischemia in utero. Two hours later, the treatment group (n = 5) received a continuous infusion of NG-nitro-L-arginine, a competitive inhibitor of NO synthase, whereas the control group (n = 6) received PBS. Changes in concentration of oxidized cytochrome oxidase, cortical impedance, and electrocortical activity were observed for 3 d. A delayed increase in cortical impedance of similar magnitude and duration commenced at 14 ± 4 h in the control and at 15 ± 3 h in the treatment groups. The progressive decrease in oxidized cytochrome oxidase signal, by -2.2 ± 0.2 µmol/L in the control and -2.0 ± 0.4 µmol/L in the treatment group at 72 h postischemia, was similar in both groups. In both groups, delayed cortical seizures were indicated by intense low-frequency electrocortical activity. In the treatment group, duration of cortical seizures was increased and the intensity of the final electrocortical activity was more depressed (-19 ± 1 dB versus -10 ± 2 dB). The results indicate that after cerebral ischemia in fetal sheep, NO synthase inhibition does not ameliorate the delayed disruptions in cerebral energetics. However, the effect of NO synthase inhibition on delayed cortical seizures may improve our understanding of the role of NO during this phase.
Similar content being viewed by others

Main
Severe transient cerebral ischemia in fetal sheep is followed by a phase of delayed cerebral injury during which impaired cellular membrane function can be detected as an increase in CI and seizures(1). These changes are accompanied by a progressive decrease in the near-infrared optical signal attributed to [CytO2], which may reflect a disruption in mitochondrial function and oxygenation(2). Delayed impairment in cerebral energy metabolism is an important feature of hypoxic-ischemic cerebral damage in the developing brain. It has been observed in rat pups, newborn piglets, and asphyxiated infants with 31P magnetic resonance spectroscopy (MRS) as a delayed decrease in the concentration of high-energy phosphates(3–5). Although a cascade of biochemical events after perinatal cerebral ischemia culminates in delayed disruptions in cerebral energetics, the role of NO has not been clarified(6).
We have previously shown that NOS inhibition reduces the delayed increase in cerebral perfusion that accompanies the delayed increase in CI(7). Further, microdialysis studies in fetal sheep have shown a delayed increase in the extracellular concentration of citrulline, reflecting an increase in the production of NO from L-arginine by NOS(8). NO can be highly neurotoxic: in combination with superoxide radicals, peroxynitrite is thought to mediate the neurotoxic effects of glutamate(9). Further, NO is particularly toxic to mitochondrial enzymes, and the inhibitory effect exerted by NO on mitochondrial respiration may well contribute to the cytotoxic effects of NO(10).
The present study investigated the hypothesis that inhibition of NO production after severe transient cerebral ischemia in fetal sheep ameliorates the delayed impairment of cerebral energetics. To determine the contribution of delayed production of NO to the delayed disruptions in cerebral energetics, the effect of L-NNA on delayed changes in CI, [CytO2], and ECoG was investigated in late gestation fetal sheep. These variables are all measures of energy metabolism and reflect different aspects of membrane function.
METHODS
Surgical procedure. Nineteen singleton Romney Suffolk fetal sheep of known gestational age (119-133 d) were operated on by sterile techniques while they were anesthetized with 2% halothane/oxygen. The general experimental approach has been described previously(1). Briefly, the 90-min procedure involved externalization of the fetal head, neck, and forelimbs and placement of polyvinyl catheters into the axillary arteries and amniotic cavity for the measurement of mean arterial blood pressure and into the umbilical vein for drug administration. Through burr holes, three pairs of shielded stainless steel electrodes were placed on the dura overlying the parasagittal cortex for two pairs of ECoG electrodes (at 5 and 15 mm anterior and 10 mm lateral to the bregma) and a third pair of stimulating electrodes to measure CI (10 mm anterior and 15 mm lateral to the bregma).
Cerebral blood supply was confined to the carotid arteries by ligation of the vertebro-occipital anastomoses and, on each carotid artery, a double balloon inflatable cuff was placed. Near-infrared light was transmitted from laser diodes at four wavelengths (774, 826, 844, and 910 nm) and carried to and from the head via fiber-optic bundles. Optic prisms at the ends of the bundles were surgically fixed with dental cement to each side of the skull at least 3.5 cm apart. The fetus was then returned to the uterus, the connecting lines were externalized through a uterine and maternal lateral skin incision, and the muscle and skin layers were closed. After surgery, ewes were housed in metabolic cages at a constant temperature (16°C) and humidity (50%) and given free access to food and water. Antibiotics (gentamicin 80 mg i.v. to the fetus and penicillin 500 mg intramuscularly to the ewe) were administered daily for 3 d after surgery.
NIRS. The study used a commercial near-infrared spectrophotometer (NIR 500 Hamamatsu Phototonics KK, Hamamatsu City, Japan). The technique depends on the transmission of near-infrared light through living tissue and its characteristic absorbance by the three chromophores oxyhemoglobin, deoxyhemoglobin, and CytO2(11,12). Changes in the chromophore concentration can be calculated from the changes in light absorption by use of a modification of the Beer-Lambert law, which describes optical absorption in a highly scattering medium(13).
Experimental protocol. Fetuses whose arterial blood gases and lactate were normal 2 d after surgery were entered into the study (Po2 >2.27 kPa, pH >7.32, lactate <1.2 mmol/L). NIRS, CI, and filtered ECoG recordings were commenced 12 h before the ischemic insult to obtain a baseline. Transient cerebral ischemia was induced by 30-min inflation of the bilateral carotid cuffs with saline and confirmed by an isoelectric ECoG and an increase in CI.
Two hours postischemia, fetuses were randomly assigned to the control or treatment group. The treatment group received a continuous infusion of L-NNA into the umbilical vein at a dose of 50 mg/h for the first 4 h followed by 20 mg/h over the subsequent 3 d, a dose known to inhibit endothelial NOS(7).
Arterial oxygen saturation, Po2, Pco2, lactate, glucose, and Hb were measured before and immediately after the end of the occlusion and at frequent intervals during the study period. Three days postischemia, ewes were killed with an overdose of pentobarbital. All studies were approved by the Animal Ethical Committee of the University of Auckland.
Data collection. NIRS recordings of the changes in [CytO2] were displayed on-line every 30 s during the study period and were recorded on magnetic disks for later analysis.
ECoG was recorded continuously, and the intensity spectra were obtained by real-time spectral analysis as previously described. The ECoG signal was amplified 10 000 times, low pass filtered at 30 Hz, and sampled at 256 Hz. An eighth-order Butterworth low-pass filter was used with a cutoff frequency set with the -3-dB point at 30 Hz. A four-electrode technique was used to extract spectral ECoG (frequency) and CI signal from the ECoG. Increases in CI are associated with a decrease in the extracellular space that occurs concomitantly with cytotoxic edema(1).
Data analysis and statistics. For analysis of the NIRS variables, the changes in the chromophore concentration were calculated from alterations in optical attenuation by least squares multilinear regression using an algorithm that utilized accurate component spectra and took account of wavelength scattering(14). The distance between the optodes was determined during surgery and confirmed at postmortem with measuring calipers.
ECoG intensity data were log-transformed, and the onset of epileptiform activity was indicated by the development of intense (>5 dB) low-frequency activity and was confirmed by inspection of the chart recorder(15,16). Descriptive measurements were made on data that had been median filtered over 500 s. Changes in the variables [CytO2], CI, and ECoG for each fetus were averaged over 6-h time periods throughout the study period. Changes in the averaged data within each group were determined by repeated measure ANOVA with time as the repeated measure, and difference between the groups was determined by ANOVA and Student-Newman-Keuls multiple comparisons test. Off-line signal analyses were performed by Viewdac Data Acquistion, version 2.1 (Keithley Data Acquisition Division, Keithley Instruments, Inc., Taunton, MA), and statistics were calculated by Sigmastat Statistical Analysis System, version 1.02 (Jandel Scientific, Erkrath, Germany). Significant results (p value of < 0.05) are presented as mean ± SEM. The end of the carotid artery occlusion is referred to as time zero.
RESULTS
Study groups. Nineteen subjects were investigated of which eight were rejected, three because of premature labor and five who died before the end of the study period. Two sham-operated animals treated with L-NNA did not show changes in CI, [CytO2], or ECoG intensity during the study period. There was no difference in mortality between the control and treatment group. Eleven fetuses were allocated to control (n = 5) and treatment (n = 6) groups and were similar for gestational age (127 ± 1.2 d), weight (3.9 ± 0.2 kg), and head circumference (5.8 ± 0.1 cm). As previously published, there was no significant difference in arterial oxygen saturation, Po2, Pco2, glucose, and lactate between the control and treatment group throughout the study period(7). The effect of L-NNA on changes in cerebral blood volume measured by NIRS and histologic outcome has been previously reported(7).
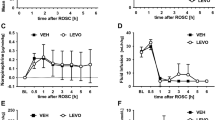
Effects of L-NNA on [CytO2]. Figure 1 shows the changes in [CytO2] signal in the control and treatment groups. In the control group, a delayed decrease in [CytO2] signal commenced 54 h postischemia and reached a minimum of -2.2 ± 0.2 µmol/L at 72 h. In the treatment group, a progressive decrease in [CytO2] signal also commenced at 54 h and reached a minimum of -2.0 ± 0.4 µmol/L at the end of the study period.

Changes in [CytO2] in µmol/L in control and L-NNA-treated group before and after cerebral ischemia. The figure shows the changes in the optical signal for [CytO2] as measured by NIRS in µmol/L during and after cerebral ischemia in control (○) and L-NNA-treated fetuses (·). In both groups, there was a progressive delayed decrease in [CytO2] that commenced at 54 h postischemia and continued throughout the remainder of the study period. Each symbol represents the mean ± SEM of averaged data for each group, and the cross-hatched bar represents the period of L-NNA administration in the treatment group.
Effect of L-NNA on ECoG. The effect of L-NNA on ECoG intensity and delayed cortical seizures is shown in Figures 2 and 3. Figure 2 shows the ECoG intensity in the control and the L-NNA-treated group during and after cerebral ischemia. In both groups, ECoG intensity was depressed during and after ischemia, followed by a slow recovery. A delayed increase in ECoG intensity toward baseline between 12 and 36 h represents the onset of intense low-frequency ECoG associated with cortical seizures shown in Figure 3. The onset of seizures was similar in both groups, although the delayed cortical seizures persisted longer in the treatment group compared with the control group (p < 0.05). As previously reported, in both groups ECoG intensity was depressed below preischemic baseline at the end of the study period and was more depressed in the treatment (-19 ± 1 dB) compared with the control group (-10 ± 2 dB) (p < 0.05)(7).

Changes in ECoG intensity in control and L-NNA-treatment group during and after cerebral ischemia. The figure shows the changes in ECoG intensity during and after cerebral ischemia in control (○) and L-NNA-treated fetuses (·). In both groups, ECoG is depressed during and after the insult. Several hours after the insult, there is an increase in intensity that represents the intense low-frequency epileptiform activity associated with cortical seizures. Greater depression of ECoG intensity by the end of the study period was evident in the treated animals. Each symbol represents the mean ± SEM of averaged data for each group, and the cross-hatched bar represents the period of L-NNA administration in the treatment group. ★Represents significant differences between groups (p < 0.05).

Seizure count in control and L-NNA-treatment group during and after cerebral ischemia. The figure shows the seizure count (seizures per hour) from 2 h after cerebral ischemia in control (○) and L-NNA-treated fetuses (·). Cortical seizures accompanied the delayed cerebral injury as represented by the delayed increase in CI. Cortical seizures persisted over a longer duration in the treated fetuses compared with the control group (p < 0.05). Each symbol represents the mean ± SEM of averaged data for each group, and the cross-hatched bar represents the period of L-NNA administration in the treatment group. ★Represents significant differences between groups (p < 0.05).
Effect of L-NNA on changes in CI. Figure 4 shows the changes in CI in the control and treatment groups throughout the study period. There were two phases of increased CI above baseline: 1) during ischemia and 2) commencing at 14 ± 4 h postischemia in the control group and at 15 ± 3 h in the treatment group. As previously reported, there was no difference between the groups in the magnitude of the increase in CI during the ischemia, recovery CI at 2-6 h, or the magnitude of the delayed increase in CI. However, the delayed increase in CI peaked 6 h later in the L-NNA group compared with the control group(7).

Changes in CI in control and L-NNA-treated group before and after cerebral ischemia. The figure shows the changes in CI during and after cerebral ischemia in control (○) and L-NNA-treated fetuses (·). In both the treatment and the control group, CI increased during the insult and recovered toward baseline with no difference in magnitude at 2-6 h. A delayed increase in CI commenced later and was of equal magnitude in both groups. Each symbol represents the mean ± SEM of averaged data for each group, and the cross-hatched bar represents the period of L-NNA administration in the treatment group.
DISCUSSION
The results of the present study indicate that after cerebral ischemia, inhibition of NO production does not ameliorate the delayed impairment of cellular energy metabolism as measured by CI or the delayed decrease in [CytO2] signal. However, the effect of NOS inhibition on these measures as well as on ECoG and delayed seizure activity contributes to our understanding of the role of NO in the pathogenesis of delayed cerebral injury. NOS inhibition was related to a depressed ECoG intensity, particularly marked by the end of the study period, and an increased duration of delayed seizure activity. These results will be discussed in the light of previous findings that in fetal sheep, NOS inhibition reduces cerebral perfusion during the development of delayed cerebral injury and increases the extent of neuronal injury(7).
The biphasic increase in CI that follows transient cerebral ischemia in fetal sheep has been used to describe the phases of cerebral injury(1). An increase in CI reflects cytotoxic edema that occurs as a consequence of failure of ionic pumps to maintain homeostasis across cellular membranes. The delayed increase in CI is thought to reflect the delayed decrease in high-energy phosphates measured by 31P MRS in asphyxiated infants(17). In fetal sheep, the delayed increase in CI is associated with a severe global ischemic insult lasting for >20 min and is inevitably associated with the development of a significant degree of cerebral injury(18).
A progressive decrease in [CytO2] measured by NIRS has been previously described in fetal sheep and neonatal piglets after severe transient cerebral ischemia(2,19). The continuous measurement of changes in [CytO2] in vivo may yield important information about the mechanism and timing of changes in mitochondrial oxygenation. The accuracy of NIRS in measuring these changes in complex and requires precise absorption spectra, validity of the Beer-Lambert relationship, and a constant optical path length throughout the experimental period. CytO2 spectra have been obtained from brains of rats after replacing the blood with a fluorocarbon substitute(20). The validity of the Beer-Lambert law has been confirmed by theoretical modeling and experimental measurement of light in scattering media; however, it depends on a constant optical path length throughout the study period(21).
The changes in the optical properties of the immature sheep brain after cerebral ischemia are unknown. Surgical fixation of the optodes to the fetal skull, which at this gestational age has fused sutures, eliminated any alteration in optical path length due to movement. However, changes in optical path length have been observed during the delayed energy failure after cerebral hypoxia-ischemia in newborn piglets(22). An increase in mean optical pathway and a reduction in the random distribution of residuals of the multilinear least squares fits of the pure chromophores may reflect changes in cell volume due to alterations in cerebral water diffusion and cellular depolarization or a more general effect after apoptotic cell death. Nevertheless, the similar time scale of the progressive decrease in [CytO2] measured by NIRS and the decrease in [ATP] measured by 31P MRS throughout the period of delayed energy failure after hypoxia-ischemia in the neonatal piglet suggests that the change in [CytO2] reflects alterations in cerebral energetics(19).
The effect of NOS inhibition on cerebral energetics after perinatal cerebral ischemia has not been previously shown. NO causes cell death by inhibiting mitochondrial respiration (complex I and II, acitonase), glycolysis [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], and DNA synthesis (ribonucleotide reductase). NO causes DNA damage that may lead to energy depletion due to activation of poly(ADP-ribose) synthase(10,23). NO also causes free radical damage by the formation of peroxynitrite, hydroxyl radical, and nitrogen dioxide(24). However, the vascular endothelial production of NO improves ischemic blood flow and is neuroprotective(25–27). In fetal sheep, L-NNA has been shown to inhibit the endothelial isoform of NOS, although inhibition of neuronal or inducible NOS has not been shown(7).
In fetal sheep, L-NNA markedly compromises the delayed increase in cerebral perfusion(7). The effect of NOS inhibition on cerebral hemodynamics may limit any beneficial effect incurred by reducing NO production in neurons, microglia, and invading macrophages after induction of inducible NOS(28). Further studies need to be performed to investigate the effect on the delayed disruptions in cerebral energetics of NOS inhibitors that specifically inhibit n-NOS and do not compromise delayed increases in cerebral perfusion(29).
Treatment with L-NNA was associated with prolongation of the delayed cortical seizures and marked depression of final ECoG intensity. In the term infant, delayed seizures are an important feature of hypoxic-ischemic encephalopathy and are associated with a particularly poor neurologic outcome(30). Control of the extent and duration of seizures has been shown to be beneficial to long-term outcome(31). Seizures associated with perinatal hypoxia-ischemia are thought to be predominantly mediated by excitatory amino acids, in particular glutamate, acting on the N-methyl-D-aspartate (NMDA) glutamate receptor(32). In fetal sheep, a massive delayed increase in glutamate concentration has been measured in the brain extracellular fluid over the time course of the delayed cortical seizures(8). Seizures are suppressed by MK-801, an NMDA-glutamate receptor antagonist, and this effect also is associated with significant neuroprotection(33).
The effect of NOS inhibition on the delayed seizures associated with hypoxia-ischemia has not been specifically investigated, and in epileptogenesis in general the results remain contradictory(34). NOS inhibitors have been shown to reduce the dose of NMDA necessary to produce convulsions, indicating that endogenous NO may play the role of an anticonvulsant substance in NMDA-induced seizures(35). NOS inhibition has also been shown to prolong the duration of seizure activity in convulsions induced by systemic or intracerebroventricular injection of NMDA into mice(36). However, L-arginine, the precursor of NO, induces seizures when administered intracerebrally in adult rats(37). The results of the present study may imply that endogenous NO has an anticonvulsant effect on the delayed seizures associated with ischemic brain injury in fetal sheep.
The effect of L-NNA on seizure activity may also reflect the reduced cerebral perfusion known to be induced by L-NNA over a similar time period. NO has been shown to couple the increased cerebral energy requirements with cerebral vasodilation during seizures(38). Limitation of this increased cerebral perfusion during seizures may promote further cerebral ischemia and increased seizures.
The effect of L-NNA on final ECoG intensity after cerebral ischemia has not been previously shown. In studies in fetal sheep, final ECoG intensity at 3-4 d postischemia is related to histologic outcome(15). However, in the present study, final ECoG intensity in the treatment group is depressed to an extent that cannot be explained by differences in histologic outcome alone(7). NO, released basally and upon activation of NMDA and glutamate receptors, has been suggested to be an important transmitter within the CNS(39). In the normal adult brain, NOS inhibition has been shown to significantly reduce sound-evoked ECoG arousal response, ECoG, and EEG power(40). Our findings suggest that NOS inhibition also markedly depresses ECoG in the immature brain after ischemia. This may suggest a role for basal NO release in the recovery of brain activity after ischemia. However, the effect may be related to the reduced cerebral perfusion observed in the treatment group by the end of the study period.
In summary, NOS inhibition has minimal effects on cerebral energetics after transient cerebral ischemia in fetal sheep as measured by the biphasic increase in CI and the progressive decrease in [CytO2]. However, the effects on ECoG were significant and suggest a role for NO in the recovery of brain activity from ischemia and the generation of delayed seizures.
Abbreviations
- NIRS:
-
near-infrared spectroscopy
- CI:
-
cortical impedance
- ECoG:
-
electrocortical activity
- [CytO2]:
-
concentration of oxidized cytochrome oxidase
- NOS:
-
nitric oxide synthase
- NO:
-
nitric oxide
- L-NNA:
-
NG-nitro-L-arginine
References
Williams CE, Gunn AJ, Gluckman PD 1991 Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 22: 516–521
Marks KA, Mallard EC, Roberts I, Williams CE, Sirimanne ES, Johnston B, Gluckman PD, Edwards AD 1996 Delayed vasodilation and altered oxygenation after cerebral ischemia in fetal sheep. Pediatr Res 39: 48–54
Roth SC, Edwards AD, Cady EB, Delpy DT, Wyatt JS, Azzopardi D, Baudin J, Townsend J, Stewart AL, Reynolds EO 1992 Relation between cerebral oxidative metabolism following birth asphyxia, and neurodevelopmental outcome and brain growth at one year. Dev Med Child Neurol 34: 285–295
Blumberg RM, Cady EB, Wigglesworth JS, McKenzie JE, Edwards AD 1997 Relation between delayed impairment of cerebral energy metabolism and infarction following transient focal hypoxia-ischaemia in the developing brain. Exp Brain Res 113: 130–137
Lorek A, Takei Y, Cady EB, Wyatt JS, Penrice J, Edwards AD, Peebles DM, Wylezinska M, Owen-Rees H, Kirkbridge V, Cooper C, Aldridge RF, Roth SC, Brown GC, Delpy DT, Reynolds EOR 1994 Delayed (“secondary”) cerebral energy failure following acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by 31P magnetic resonance spectroscopy. Pediatr Res 36: 699–706
Vannucci RC 1993 Mechanisms of perinatal hypoxic-ischemic brain damage. Semin Perinatol 17: 330–337
Marks KA, Mallard CE, Roberts I, Williams CE, Gluckman PD, Edwards AD 1996 Nitric oxide synthase inhibition attenuates delayed vasodilation and increases injury after cerebral ischemia in fetal sheep. Pediatr Res 40: 185–191
Tan WK, Williams CE, During MJ, Mallard CE, Gunning MI, Gunn AJ, Gluckman PD 1996 Accumulation of cytotoxins during the development of seizures and edema after hypoxic-ischemic injury in late gestation fetal sheep. Pediatr Res 39: 791–797
Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH 1991 Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 88: 6368–6371
Zhang J, Dawson VL, Dawson TM, Snyder SH 1994 Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science 263: 687–689
Jobsis FF, Keizer JH, LaManna JC, Rosenthal M 1977 Reflectance spectrophotometry of cytochrome aa3 in vivo. J Appl Physiol 43: 858–872
Reynolds EO, Wyatt JS, Azzopardi D, Delpy DT, Cady EB, Cope M, Wray S 1988 New non-invasive methods for assessing brain oxygenation and haemodynamics. Br Med Bull 44: 1052–1075
Edwards AD 1995 Near infrared spectroscopy. Eur J Pediatr 154( suppl 3): S19–S21
Matcher SJ, Elwell CE, Cooper CE, Cope M, Delpy DT 1995 Performance comparison of several published tissue near-infrared spectroscopy algorithms. Anal Biochem 227: 54–68
Williams CE, Gunn AJ, Synek B, Gluckman PD 1990 Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res 27: 561–565
Williams CE, Gluckman PD 1990 Real-time spectral intensity analysis of the EEG on a common microcomputer. J Neurosci Methods 32: 9–13
Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, Hope PL, Hamilton PA, Reynolds EO 1989 Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr Res 25: 445–451
Williams CE, Gunn AJ, Mallard C, Gluckman PD 1992 Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann Neurol 31: 14–21
Cooper CE, Springett R 1997 Measurement of cytochrome oxidase and mitochondrial energetics by near-infrared spectroscopy. Philos Trans R Soc Lond B Biol Sci 352: 669–676
Cooper CE, Cope M, Quaresima V, Ferrari M, Nemoto E, Springett R, Matcher S, Amess P, Penrice J, Tyszczuk L, Wyatt J, Delpy DT 1997 Measurement of cytochrome oxidase redox state by near infrared spectroscopy. Adv Exp Med Biol 413: 63–73
Cope M, Delpy DT, Reynolds EOR, Wray S, Wyatt J, van der Zee P 1988 Methods of quantitating cerebral near infrared spectroscopy data. Adv Exp Med Biol 222: 183–189
Hiraoka M, Firbank M, Essenpreis M, Cope M, Arridge SR, van der Zee P, Delpy DT 1993 A Monte Carlo investigation of optical pathlength in inhomogenous tissue and its application to near-infrared spectroscopy. Phys Med Biol 38: 1859–1876
Dawson TM, Dawson VL, Snyder SH 1993 Nitric oxide as a mediator of neurotoxicity. NIDA Res Monogr 136: 258–271
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA 1990 Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87: 1620–1624
Faraci FM, Brian JEJ 1994 Nitric oxide and the cerebral circulation. Stroke 25: 692–703
Nishikawa T, Kirsch JR, Koehler RC, Bredt DS, Snyder SH, Traystman RJ 1993 Effect of nitric oxide synthase inhibition on cerebral blood flow and injury volume during focal ischemia in cats. Stroke 24: 1717–1724
Prado R, Watson BD, Wester P 1993 Effects of nitric oxide synthase inhibition on cerebral blood flow following bilateral carotid artery occlusion and recirculation in the rat. J Cereb Blood Flow Metab 13: 720–723
Lees GJ 1993 The possible contribution of microglia and macrophages to delayed neuronal death after ischemia. J Neurol Sci 114: 119–122
Iadecola C, Zhang F, Xu X 1995 Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol 268:R286–R292
Finer NN, Robertson CM, Richards RT, Pinnell LE, Peters KL 1981 Hypoxic-ischemic encephalopathy in term neonates: perinatal factors and outcome. J Pediatr 98: 112–117
Vannucci RC 1990 Current and potentially new management strategies for perinatal hypoxic-ischemic encephalopathy. Pediatrics 85: 961–968
Hill A, Volpe JJ 1985 Pathogenesis and management of hypoxic-ischemic encephalopathy in the term newborn. Neurol Clin 3: 31–46
Tan WK, Williams CE, Gunn AJ, Mallard CE, Gluckman PD 1992 Suppression of postischemic epileptiform activity with MK-801 improves neural outcome in fetal sheep. Ann Neurol 32: 677–682
Kirkby RD, Carroll DM, Grossman AB, Subramaniam S 1996 Factors determining proconvulsant and anticonvulsant effects of inhibitors of nitric oxide synthase in rodents. Epilepsy Res 24: 91–100
Buisson A, Lakhmeche N, Verrecchia C, Plotkine M, Boulu RG 1993 Nitric oxide: an endogenous anticonvulsant substance. Neuroreport 4: 444–446
De Sarro GB, Donato Di Paola E, De Sarro A, Vidal MJ 1991 Role of nitric oxide in the genesis of excitatory amino acid-induced seizures from the deep prepiriform cortex. Fundam Clin Pharmacol 5: 503–511
Mollace V, Bagetta G, Nistico G 1991 Evidence that L-arginine possesses proconvulsant effects mediated through nitric oxide. Neuroreport 2: 269–272
Montecot C, Borredon J, Seylaz J, Pinard E 1997 Nitric oxide of neuronal origin is involved in cerebral blood flow increase during seizures induced by kainate. J Cereb Blood Flow Metab 17: 94–99
Snyder SH 1992 Nitric oxide: first in a new class of neurotransmitters. Science 257: 494–496
Pelligrino DA, Gay RL, Baughman VL, Wang Q 1996 NO synthase inhibition modulates NMDA-induced changes in cerebral blood flow and EEG activity. Am J Physiol 271:H990–H995
Author information
Authors and Affiliations
Additional information
Supported by The Wellcome Trust, No. 038916, National Institutes of Health, and the Health Research Council of New Zealand.
Rights and permissions
About this article
Cite this article
Marks, K., Mallard, C., Roberts, I. et al. Nitric Oxide Synthase Inhibition and Delayed Cerebral Injury after Severe Cerebral Ischemia in Fetal Sheep. Pediatr Res 46, 8–13 (1999). https://doi.org/10.1203/00006450-199907000-00002
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199907000-00002
