
Abstract
Acylcarnitines are important diagnostic markers for inborn errors of fatty acid oxidation, but their analysis in body fluids may not always be reliable. Recently, disease-specific acylcarnitine profiles generated by cultured skin fibroblasts were reported to facilitate the diagnosis by localizing a specific enzymatic defect in the mitochondrial β-oxidation pathway. Using a novel methodologic approach, fibroblasts from 16 patients with inborn errors of fatty acid oxidation and 13 control subjects were preincubated with L-[3H]carnitine to label the intracellular carnitine pool. Cells were subsequently incubated with unlabeled palmitic acid and, after methanol extraction of cells and media, labeled free carnitine and acylcarnitines were analyzed by radio-HPLC. Quantitation was based on the integrated radioactivity of individual peaks relative to the total radioactivity recovered. In control cell lines, all saturated acylcarnitines were detected, and reference values were established. With the exception of one cell line deficient in electron transfer flavoprotein, all mutant cell lines showed abnormal and disease-specific relative concentrations of acylcarnitines. Advantages of the method include use of a small number of cells, no need for trypsinization and permeabilization of cells before incubation, simple extraction without purification of the specimen before HPLC, and relatively inexpensive equipment. The method allows a focused approach to the subsequent, more laborious confirmation of a particular disease by direct enzymatic and/or molecular analysis. It remains to be established whether the method can replace widely used global measurements of fatty acid oxidation rates in vitro that do not provide specific information about the enzyme deficiency involved.
Similar content being viewed by others

Main
In recent years, an increasing number of patients with inborn errors of mitochondrial fatty acid oxidation have been described. At least 18 different enzyme defects are known to date. These diseases represent one of the most common groups of inborn errors of metabolism and are life-threatening, particularly in infancy. Unexpected deaths have occurred in approximately 20% of affected families (for review, seeRef. 1). Clinical features include a recurrent Reye-like syndrome with fatty liver, hepatic failure, hypoketotic hypoglycemic coma, cardiomyopathy, muscle hypotonia, and chronic encephalopathy. There is considerable overlap of symptoms among the different enzyme deficiencies, so that a specific diagnosis cannot usually be assigned from the clinical phenotype alone.
The timely diagnosis of inborn errors of fatty acid oxidation is of paramount importance because life-threatening metabolic crises can often be prevented by appropriate dietary intervention. These diseases may escape routine metabolic screening because characteristic metabolites may not be excreted in the symptom-free interval(2). Because most inborn errors of fatty acid oxidation are associated with alterations in carnitine metabolism resulting in increased concentrations of characteristic carnitine esters (AC) in body fluids and tissues, AC are important diagnostic markers for these diseases. However, it is recognized that AC concentrations in body fluids depend upon the metabolic state and the carnitine status of the patient(3). These concentrations may be extremely low if patients are in a stable metabolic condition and if they are not supplemented with L-carnitine. Therefore, as a diagnostic tool, AC analysis in body fluids may not always be reliable.
Recently, disease-specific AC profiles generated by cultured skin fibroblasts(4,5) or isolated peripheral blood cells(6) from patients with inborn errors of fatty acid oxidation were reported. Such in vitro methods may facilitate the diagnosis of inborn errors of fatty acid oxidation by localizing a specific enzymatic defect of the mitochondrial β-oxidation pathway. Two different methodologies have been used. Fibroblasts are incubated with fatty acids labeled either with stable isotopes(5,7) or with radioisotopes(4), and the labeled AC are subsequently analyzed using tandem mass spectrometry or radio-HPLC, respectively. Whereas the former method requires expensive equipment, the latter method requires cell permeabilization and extensive purification procedures to eliminate radiolabeled metabolites other than AC from the specimen. We circumvented these problems by preincubating fibroblasts with L-[3H]carnitine to label the intracellular carnitine pool. Cells were subsequently incubated with unlabeled palmitic acid as substrate resulting in the production of AC labeled at the carnitine moiety. These were readily analyzed by radio-HPLC after simple methanol extraction.
METHODS
Patients' cells. Fibroblasts were obtained from skin biopsies of 13 subjects who had no evidence of an inborn error of fatty acid oxidation by in vitro oxidation studies (control subjects), and of 16 patients with inborn errors of fatty acid oxidation (see Table 1), including deficiencies of CPT I, CPT II (early onset), VLCAD, TFE, LCHAD, MCAD, SCAD, ETF-α, and ETF-DH. Diagnoses were established by direct enzyme and/or DNA mutation (LCHAD, MCAD, SCAD) analysis. One patient with MCAD deficiency was diagnosed postmortem by the current method. He was subsequently found to be homozygous for the common A985G mutation. Two other patients with MCAD deficiency were homozygous for the same mutation, and one additional patient was a compound heterozygote with A985G and G799A mutations. The patient with LCHAD deficiency was found to have a reduced activity of LCHAD with normal activities of mitochondrial long chain enoyl-CoA hydratase and long chain 3-ketothiolase. He was homozygous for the G1528C mutation. The patient with TFE deficiency was found to have reduced activities of all three components of this enzyme (our unpublished data). The patient with SCAD deficiency was a compound heterozygote with two unique mutant alleles(8). All patients with ETF-α/ETF-DH deficiency had severe glutaric aciduria type II with neonatal onset. The study was approved by the Institutional Review Board of Louisiana State University Medical Center of New Orleans.
Incubation of fibroblasts. Fibroblasts were grown to confluence in 25-cm2 tissue culture flasks (Corning Inc.) using Dulbecco's modified Eagle's medium (GIBCO BRL, Gaithersberg, MD) supplemented with 10% fetal bovine serum. The cells were then transferred to 60-mm Petri dishes and grown to approximately 50% confluence over 2-3 d. At this point, the medium was changed to Dulbecco's modified Eagle's medium/fetal bovine serum containing 1 µCi/mL L-[methyl-3H]carnitine hydrochloride (specific activity 78 Ci/mmol; Amersham Corp., St. Louis, MO). Upon reaching confluence (3-6 d), the cells were rinsed twice with PBS and incubated at 37°C, 5% CO2 for 2 h in 2 mL of "palmitic acid medium." This medium had been prepared by evaporating 300 µL of a 2.2 mM palmitic acid (Sigma Chemical Co.)-ethanol solution to dryness and complexing it with 300 µL of 1% essential fatty acid-free BSA (Sigma Chemical Co.) in Krebs-Ringer's bicarbonate (pH 6.9) using constant agitation in a 37°C waterbath for 30 min with intermittent vortexing. Krebs-Ringer's bicarbonate (pH 6.9) was added to yield 6 mL of palmitic acid medium. After the incubation, the cells were removed by scrapping, and the cells plus medium were frozen.
Extraction of fibroblasts and media. The cell suspension was thawed, mixed with 5 volumes of methanol, vortexed for 2 min, and then centrifuged at 750 × g. The supernatant was removed, and the pellet was washed three times with 2 mL of methanol. Supernatant and washes were combined and evaporated to dryness at 37°C under nitrogen. One milliliter of methanol was added, and the solution was vortexed and then recentrifuged. The supernatant was removed, and the second precipitate washed twice with 2 mL of methanol. Supernatant and washes were combined and evaporated as before, dissolved in 100 µL of methanol:water 1:1(vol/vol), and filtered. Recovery of total radioactivity from fibroblasts and media was greater than 90%.
Radio-HPLC. Labeled free carnitine and AC were analyzed by radio-HPLC as described previously(9).3 H-Labeled free carnitine and individual AC were identified by comparing their relative retention times with those of authentic standards. Standards of labeled short and medium chain (C2-C10)-AC were prepared from unlabeled compounds (purchased from Sigma Chemical Co. or synthesized according to Bohmer and Bremer(10) using an enzymatic radioisotopic exchange reaction(9).3 H-Labeled dodecanoyl (C12)- and tetradecanoyl(C14)-carnitine were synthesized by incubating the corresponding coenzyme A esters (Sigma Chemical Co.) with L-[3H]carnitine, N-ethylmaleimide (Sigma Chemical Co.), and carnitine acetyl transferase (Boehringer Mannheim). 3H-Labeled hexadecanoyl(C16)-carnitine was synthesized from hexadecanoyl-CoA (Sigma Chemical Co.) using CPT II (kindly provided by Dr. G. A. Cook, Department Pharmacology, College of Medicine, University of Tennessee).3 H-Labeled 3-OH- and 3-oxo-C16-carnitine were synthesized from the corresponding acyl-CoA esters using the same enzyme. The latter were prepared from hexadecanoyl-CoA using acyl-CoA oxidase, crotonase and 3-OH-acyl-CoA dehydrogenase (all from Sigma Chemical Co.). They were quantitatively resolved by HPLC. A standard mixture of C2-C16-AC was run before and after each series of samples. The variability of the retention times between these two standard runs generally did not exceed±0.1 min. The retention times of C14-, 3-OH-C16-, 3-oxo-C16-, and C16-AC were 44.10, 44.77, 44.80, and 46.40 min, respectively.
Quantitation was based on the integrated radioactivity of individual peaks. Results were expressed as percent of the sum of integrated radioactive peaks. They were reported by the analyzing laboratory (E.S.S., P.J.B., D.P.) without prior knowledge of the diagnosis.
RESULTS
Control cell lines. In all control cell lines, free carnitine and the following carnitine metabolites were detected and their relative concentrations quantitated: acetyl (C2)-, propionyl (C3)-, butyryl/isobutyryl (C4)-, isovaleryl/2-methylbutyryl (C5)-, hexanoyl (C6)-, octanoyl (C8)-, decanoyl (C10)-, dodecanoyl (C12)-, and hexadecanoyl (C16)-carnitine(Fig. 1a). Tetradecanoyl (C14)-carnitine was detected in only half of the cell lines. Median and range for integrated radioactivity in individual peaks of control fibroblasts are presented in Table 1.

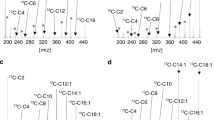
HPLC-chromatograms of3 H-labeled free carnitine (FC) and carnitine esters of different chain lengths (C2-C16) accumulated in cultured skin fibroblasts and media. (a) control; (b) CPT I deficiency; (c) CPT II deficiency; (d) VLCAD deficiency;(e) LCHAD deficiency; (f) MCAD deficiency; (g) SCAD deficiency; (h) ETF-DH deficiency. The abscissas depict retention times. *Presumed 3-OH-C16-AC.
Cell lines from patients with inborn errors of fatty acid oxidation. The patient with TFE deficiency and one patient with ETF-α deficiency had normal relative concentrations of labeled C2-AC in fibroblasts. All other cell lines from patients with metabolic disease had decreased C2-AC levels (Table 1). In cells from two patients with CPT I deficiency, over 80% of the radioactivity remained in the free carnitine peak and little radioactivity was detected in AC (Fig. 1b). Cells from two patients with CPT II deficiency showed increased relative concentrations of C16-AC(Fig. 1c). Small amounts ofC2- and C3-AC were found, but no radioactivity was detected in the other carnitine esters. The VLCAD-deficient cell line showed increased relative concentrations of C14- and C16-AC (Fig. 1d). The concentration of C14-AC was higher than that found in patients with ETF-α or ETF-DH deficiency (Table 1). Cells from patients with TFE and LCHAD deficiency had increased relative concentrations of C16-AC. In addition, a characteristic compound with a retention time longer than C14- and shorter than C16-AC was noted in both cell lines(Fig. 1e). The retention time was close to that of 3-OH-C16 or 3-oxo-C16 standards. This compound was consistently detected in six separate experiments with blinded interlaboratory analysis of these two cell lines and was not present in any of the other cell lines studied. Cells from four patients with MCAD deficiency showed increased relative concentrations of C8-AC (Fig. 1f), with no overlap with control values. C14- and C16-AC were also elevated in three of these patients. The SCAD deficient cell line showed an increased relative concentration of C4-AC (retention time of butyrylcarnitine, Fig. 1g). There were two cell lines deficient for the ETF-α subunit. One of them showed relative increases in C5-AC, as well as C12-, C14-, and C16-AC, whereas the other cell line did not show any abnormalities. In both ETF-DH-deficient cell lines, C5-, C14-, and C16-AC were increased(Fig. 1h).
DISCUSSION
Our results confirm previous findings that disease-specific patterns of AC are produced by fibroblasts after 2 h of incubation with the fatty acid substrate hexadecanoic acid. The 2-h incubation period was shown previously to produce a consistent pattern of accumulated carnitine metabolites by human fibroblasts(4). Whereas C2-AC was the main carnitine metabolite generated by control fibroblasts, this carnitine ester was decreased in most abnormal cell lines reflecting a general impairment of the β-oxidation pathway and decreased rates of acetyl-CoA production.
With the exception of CPT I deficiency, all defects of long chain fatty acid oxidation were characterized by excess accumulation of C16-AC. The most prominent relative concentrations of this metabolite were found in the cell lines with CPT II deficiency. TFE and LCHAD deficiencies could easily be differentiated from the other long chain fatty acid oxidation defects by the additional production of an AC with a retention time close to that of 3-OH-C16 or 3-oxo-C16-AC. This compound most likely is 3-OH-C16-AC as has previously been observed using different methods(4,5). However, it is possible that TFE-deficient fibroblasts also accumulate hexadec-2-enoylcarnitine(4) or 3-oxo-C16-AC. VLCAD deficiency was characterized by the additional prominent elevation of C14-AC. As shown before, cell lines with CPT I deficiency produced no long or medium chain AC. In addition, in cells with this defect, the amount of radioactivity remaining in the free carnitine peak was much larger than in any other cell line. This feature, which appears to be unique in CPT I deficiency and cannot be recognized by other previously published methods, may allow the unequivocal recognition of CPT I deficiency in cultured fibroblasts.
MCAD deficiency was easily recognized by prominent C8-AC concentrations in all four cases. However, in contrast to previous reports(4,5,7), concentrations of C10-AC were not consistently increased. Our cell line with SCAD deficiency showed a prominent concentration of C4-AC, the retention time of which corresponded to butyrylcarnitine. Chromatographic separation of the two isomers of C4-AC appears to be important, because butyrylcarnitine derived from fatty acid oxidation must be differentiated from isobutyrylcarnitine, which is derived from branched chain amino acid metabolism. For example, elevated concentrations of isobutyrylcarnitine in plasma and urine have been found in patients with congenital lactic acidosis(11), whereas patients with SCAD deficiency have increased levels of butyrylcarnitine(12). Although the retention times of butyryl- and isobutyrylcarnitine are distinct under the above described HPLC conditions, an alternative solvent system(13) may be required for unequivocal identification of these isomers.
Marked biochemical heterogeneity of ETF-α and ETF-DH deficiencies has been reported(14). This may explain why one of our four patients with these defects showed no abnormality of the AC pattern produced by fibroblasts. However, this patient's phenotype was not different from that of the other three cases, where long chain AC and C5-AC were clearly elevated. The latter compounds are presumably derived from breakdown of the endogenous branched chain amino acids leucine and isoleucine. It is possible that the sensitivity of the method for the detection of ETF-α and ETF-DH deficiencies can be improved by adding branched chain amino acids as precursors to the incubation medium. The high concentrations of C5-AC and the C14/C16-AC ratio differentiate cell lines from patients with ETF-α and ETF-DH deficiencies from those with VLCAD deficiency.
Our method is fundamentally different from previously described methods that measure AC formation in vitro. In this method, the fatty acid precursor is unlabeled and the intracellular carnitine pool is labeled. Therefore, the method does not allow the direct calculation of fatty acid metabolite flux through the carnitine esterification pathways. However, our results suggest that, for diagnostic purposes, such calculations are not needed. Instead, quantitating the incorporation of labeled carnitine into the different AC fractions relative to the total labeled carnitine pool suffices for establishing a diagnosis. Additional advantages of the method are as follows. 1) Only small numbers of cells (28-cm2 dish) are needed for one experiment. This is in contrast to between 75 and 160 cm2 of attached fibroblasts used in other assays(4,7). 2) Trypsinization and permeabilization of cells before incubation with substrate are not needed; instead, cells are incubated while attached to the dish. In other studies, no labeled AC could be detected by radio-HPLC if fibroblasts were incubated for up to 2 h with labeled hexadecanoic acid and L-carnitine, unless the cells were permeabilized(4). Cell permeabilization presumably improves access of substrates, particularly L-carnitine, to β-oxidation. However, we were able to consistently detect most carnitine metabolites in control fibroblasts. This was most likely achieved by the prolonged preincubation of cells with labeled carnitine resulting in a labeled intracellular carnitine pool of high specific activity and allowing the detection of relatively low concentrations of AC formed during fatty acid oxidation. 3) Purification of the specimen before radio-HPLC is not needed, because only carnitine and carnitine metabolites are labeled and detected. This is in contrast to other methods where labeled carnitine esters need to be separated from other fatty acid-derived labeled compounds which might interfere with the chromatographic analysis(4).4) The method can be performed in any laboratory with experience in tissue culture and HPLC analysis and requires relatively inexpensive equipment.
In conclusion, preincubation of fibroblasts from patients with inborn errors of fatty acid oxidation with L-[3H]carnitine and subsequent incubation with hexadecanoic acid results in the formation of disease-specific patterns of AC. This allows a focused approach to the subsequent more laborious confirmation of a particular disease by direct enzymatic and/or molecular analysis. The described method represents a simplified approach to in vitro diagnosis of these diseases in that it requires only small amounts of cells, no pretreatment of cells, no purification or derivatization procedures before radio-HPLC, and readily available equipment. It remains to be established whether the method can replace widely used global measurements of fatty acid oxidation rates in vitro, which do not provide specific information about the enzyme deficiency involved(15,16).
Abbreviations
- AC:
-
acylcarnitine(s)
- CPT:
-
carnitine palmitoyltransferase
- VLCAD:
-
very long chain acyl-CoA dehydrogenase
- TFE:
-
trifunctional enzyme
- LCHAD:
-
long chain 3-hydroxy acyl-CoA dehydrogenase
- MCAD:
-
medium chain acyl-CoA dehydrogenase
- SCAD:
-
short chain acyl-CoA dehydrogenase
- ETF-α:
-
electron transfer flavoprotein α-subunit
- DH:
-
dehydrogenase
References
Roe CR, Coates PM 1995 Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp 1501–1533
Vockley J 1994 The changing face of disorders of fatty acid oxidation. Mayo Clin Proc 69: 249–257
Schmidt-Sommerfeld E, Penn D, Kerner J, Bieber LL, Rossi TM, Lebenthal E 1989 Quantitation of urinary carnitine esters in a patient with medium chain acyl-coenzyme A dehydrogenase deficiency: effect of metabolic state and L-carnitine therapy. J Pediatr 115: 577–582
Pourfarzam M, Schaefer J, Turnbull DM, Bartlett K 1994 Analysis of fatty acid oxidation intermediates in cultured fibroblasts to detect mitochondrial oxidation disorders. Clin Chem 12: 2267–2275
Nada MA, Rhead WJ, Sprecher H, Schulz H, Roe CR 1995 Evidence for intermediate channeling in mitochondrial β-oxidation. J Biol Chem 270: 530–535
Schaefer J, Pourfarzam M, Bartlett K, Jackson S, Turnbull DM 1995 Fatty acid oxidation in peripheral blood cells: characterization and use for the diagnosis of defects of fatty acid oxidation. Pediatr Res 37: 354–360
Nada MA, Chace DH, Sprecher H, Roe CR 1995 Investigation of β-oxidation intermediates in normal and MCAD-deficient human fibroblasts using tandem mass spectrometry. Biochem Mol Med 54: 59–66
Naito E, Indo Y, Tanaka K 1990 Identification of two variant short chain acylcoenzyme A dehydrogenase alleles, each containing a different point mutation in a patient with short chain acyl-coenzyme A dehydrogenase deficiency. J Clin Invest 85: 1575–1582
Schmidt-Sommerfeld E, Zhang L, Bobrowski PJ, Penn D 1995 Quantitation of short-and medium-chain acylcarnitines in plasma by radioisotopic exchange/high-performance liquid chromatography. Anal Biochem 231: 27–33
Bohmer T, Bremer J 1986 Propionylcarnitine, physiological variations in vivo. Biochim Biophys Acta 152: 559–567
Burlina AB, Dioniso-Vici C, Bennett MJ, Gibson KM, Servideo S, Bertini E, Hale DE, Schmidt-Sommerfeld E, Sabetta G, Facchello F, Rinaldo P 1994 A new syndrome with ethylmalonic aciduria and normal fatty acid oxidation in fibroblasts. J Pediatr 124: 79–86
Bhala A, Willi SM, Rinaldo P, Bennett MJ, Schmidt-Sommerfeld E, Hale DE 1995 Clinical and biochemical characterization of short-chain acyl-CoA dehydrogenase deficiency. J Pediatr 126: 910–915
Bieber LL, Kerner J 1986 Short-chain acyl carnitines: identification and quantitation. Methods Enzymol 123: 264–276
Loehr JP, Goodman SI, Frerman FE 1990 Glutaric acidemia type II: heterogeneity of clinical and biochemical phenotypes. Pediatr Res 27: 311–315
Saudubray JM, Coude FX, Demaugre F, Johnson C, Gibson KM, Nyhan WL 1982 Oxidation of fatty acids in cultured fibroblasts: a model system for the detection and study of defects in oxidation. Pediatr Res 16: 877–881
Rhead WJ 1990 Screening for inborn errors of fatty acid oxidation in cultured fibroblasts: an overview. In: Tanaka K, Coates PM(eds) Fatty Acid Oxidation: Clinical, Biochemical and Molecular Aspects. Alan R Liss, New York, pp 365–382
Acknowledgements
The authors thank Lodewijk IJlst for synthesizing the long chain acyl-CoA esters and Pat Davillier for expert assistance in preparing this manuscript.
Author information
Authors and Affiliations
Additional information
Supported, in part, by U.S. Public Health Service Grant HD29273 and by a grant from the American Heart Association-LA Affiliate, Inc.
Rights and permissions
About this article
Cite this article
Schmidt-Sommerfeld, E., Bobrowski, P., Penn, D. et al. Analysis of Carnitine Esters by Radio-High Performance Liquid Chromatography in Cultured Skin Fibroblasts from Patients with Mitochondrial Fatty Acid Oxidation Disorders. Pediatr Res 44, 210–214 (1998). https://doi.org/10.1203/00006450-199808000-00012
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199808000-00012
This article is cited by
-
Novel ETF dehydrogenase mutations in a patient with mild glutaric aciduria type II and complex II‐III deficiency in liver and muscle
Journal of Inherited Metabolic Disease (2010)
-
Analysis of plasma free fatty acid cyanomethyl derivatives by GC-NPD for the diagnosis of mitochondrial fatty acid oxidation disorders
Chromatographia (2000)
