
Abstract
In this study, the effect of -158(C → T) (XmnI) polymorphism on the synthesis of fetal Hb and its Gγ component during the switchover from fetal to adult Hb was examined using cord blood samples from normal Caucasian term infants. The presence of -158(C→T) mutation was determined by amplification of Gγ- and Aγ-globin gene promoter fragments from the DNA isolated from cord blood samples, followed by XmnI restriction enzyme digestion. The syntheses of fetal and adult Hb in cord blood were measured by [3H]leucine incorporation in globin synthesis, separation of the globin polypeptides by HPLC, and scintillation counting of the fractions. The presence of -158(C→ T) substitution in the Gγ-globin promoter region was positively correlated with elevated synthesis of fetal Hb and itsGγ-globin component in term newborn infants and is associated with delayed switchover from fetal to adult Hb. In addition, analysis of cord blood samples from 100 normal Caucasian French Canadian term infants revealed that the frequency of -158(C → T) substitution inGγ-promoter was 0.32.
Similar content being viewed by others


Main
The changes in the expression of Hb isoforms produced during embryonic, fetal, and adult life have served as a model for understanding the regulation of human genes. The production of human Hb involves several developmental switches. These are the embryonic (ζ2ε2) to fetal(α2γ2) which begins around 6 wk of gestation(1), followed by the fetal to adult(α2β2), a switchover that begins at 28 wk and is completed at 54 wk of postconceptional age(2). Finally the proportions of Gγ- to Aγ-globin, the globin components of HbF, change from a fetal to an adult ratio (3:1 to 2:3) after 44 wk of postconceptional age(3). The most widely accepted hypothesis is that the cellular regulation of the timing of the Hb type switching is determined by the postconceptional age of the fetus(1, 2, 4, 5).
The human β gene cluster on chromosome 11p contains the functional genes ε,Gγ,Aγ,δ and β. Although a great deal is known about the structure and molecular pathology of human Hb genes, it is still not clear how their differential expression during normal development is regulated. The process of globin gene switching involves complex interactions of stage-specific transcription factors, chromosomal gene order, gene proximity to the globin locus control region, cis-acting sequences, and trans-acting factors(6). Among the genetic factors known to affect HbF production in adults are DNA sequence variations within the β-globin gene cluster. In particular, the (C → T) variation at position -158 upstream of the Gγ globin gene which is detectable by the restriction enzyme XmnI. This sequence variation has been shown to increase HbF levels in β-thalassemia and sickle cell anemia(7). Normal adults heterozygous or homozygous for the presence of this polymorphism have higher HbF levels than controls without this variation(8). An increase in HbF has also been documented in the presence of this polymorphism during pregnancy(9), Fanconi's anemia(10), and leukemia(11).
The relationship of XmnI polymorphism and HbF has been studied in adult populations; however, it is not clear if the presence (+) or absence (-) of an XmnI restriction site can modulate the level of HbF production during the switchover from HbF to HbA. When human fetal development reaches the maturation that corresponds to term gestation, the switchover from fetal to adult Hb synthesis is near the mid point of completion. However, in spite of the same postconceptional age, the relative amounts of HbF being synthesized at this time period of gestation can vary over a wide range(30-60%)(2). This dispersion of HbF production exists in these newborn infants despite the fact that they are at the same stage of their maturational development.
The considerable variation of HbF synthesis found in term infants could possibly be explained in part by the effect of the presence or absence of aXmn I polymorphism on the level of HbF expression. To test this hypothesis, the present study was carried out to determine the incidence of-158(C → T) mutation and its association with the amounts of HbF being synthesized in cord blood from term newborn infants. This study would also provide data that could be extrapolated to indicate the incidence of the C→ T substitution in a Caucasian French Canadian population.
METHODS
To evaluate the relationship between XmnI polymorphism and the synthesis of HbF and its components, cord blood samples were obtained from 59 term newborn infants delivered at St. Justine's Hospital, Montreal, who were appropriate in weight for gestational age and without known congenital anomalies. Because the level HbF synthesis is dependent on gestational age, to decrease the margin of spread in developmental age, the full-term infants included in this study were limited to only those whose gestational age was 38-41 wk. The gestational age was based on the ultrasound examination performed at 18-20 wk of pregnancy, the mother's menstrual history, and the confirmation by the infant's physical examination on the day of birth.
To obtain a better estimate of incidence of -158(C → T) polymorphism in a Caucasian French Canadian population, the sample size for the DNA analysis was enlarged by 41 randomly selected cord blood samples. Because developmental age does not affect the frequency of the mutation, there were no restrictions based on the gestational age of the infants from whom the samples were obtained. Hb synthesis was not determined in these additional samples.
Amplification and restriction analysis of theGγ- globin andAγ- globin gene promoter fragments. Cellular DNA was prepared from the buffy coat of cord blood using a DNA extraction kit (Qiagen Inc., Chatsworth, CA). DNA fragments of theGγ-globin promoter [1500-2121 nucleotides. The numbering of nucleotides was according to the sequence reported by Shen et al.(12)] The Aγ-globin promoter (6426-7051 nucleotides) was amplified by polymerase chain reaction using the following primers: hG1, 5′-CCT GCA CTG AAA CTG TTG C-3′; and hG2, 5′-AAC TGC TGA AGG GTG CTT CC-3′; for Gγ-globin promoter and hA1, 5′-TAC TGC GCT GAA ACT GTG G-3′; and hG2 forAγ-globin promoter. The PCR reactions (50 μL) contained 100 ng of DNA, 20 mM Tris-HCl, pH 8.5, 50 mM KCl, 2 mM MgCl2, 100 μM dNTPs, 2 μCi of [32P]dCTP (3000 Ci/mmol) (Amersham Canada, Mississauga, ON), 1.5 units of Taq DNA polymerase (Life Technologies, Inc., Burlington, ON) and were subjected to 35 cycles of amplification (each cycle being 94°C, 1 min; 52°C; 1 min; and 72°C, 1 min) in a Perkin-Elmer thermal cycler. An aliquot of the sample (1-5 μL) was digested with 300 U/mL XmnI (New England Biolabs, Mississauga, ON) or DraI(Life Technologies, Inc.) restriction enzymes overnight at 37°C, as instructed by the manufacturer. The DNA fragments were resolved on 6% polyacrylamide gels and the radioactive bands were visualized by autoradiography using Kodak XO-matic XAR film or by PhosphorImaging (Molecular Dynamics Inc., Sunnyvale, CA).
Quantitation of HbF synthesis and the Gγ- and Aγ- globin fractions. Incubation of the red blood cells obtained from the fresh cord blood in an amino acid mixture containing [3H]leucine for 6 h, preparation of hemolysate, globin chain separation, and quantitation by C4-reverse phase HPLC equipped with an integrator were conducted as previously described(3). After liquid scintillation counting of fractions containing separated globins, quantification of the relative amounts was performed using a computer program (Inplot 4.02, Graphpad Software Inc., San Diego, CA) that provided an integrated profile of the incorporation of[3H]leucine into the globin chains as well as their proportions. HbF is expressed as γ/(γ + β) × 100 and Gγ, asGγ/(Gγ + Aγ) × 100.
Statistical analysis. The data were analyzed in three groups; the homozygous presence of XmnI polymorphism (++), heterozygous(+-), and homozygous absence (--). The groups were compared using one-way analysis of variance and a post hoc test for linear trend. Ap ≤ 0.05 was considered as statistically significant (Instat 2, Graphpad Software Inc.).
All procedures were approved by Institutional Review Board of Hôpital Sainte-Justine.
RESULTS
Amplification of the cellular DNA isolated from cord blood usingGγ- and Aγ-specific primer pairs by PCR resulted in 621- and 624-bp DNA fragments, respectively. These DNA fragments were cloned in pGEM3 plasmid, and several plasmid recombinants were sequenced to confirm specific amplification of Gγ- and Aγ-globin promoter fragments by their respective primer pairs. After digestion withXmn I restriction endonuclease, the Gγ-globin PCR product yielded the following DNA fragments, depending on the presence or absence of XmnI recognition sequence on one (+/-), both (+/+), or none (-/-) of the diploid chromosome complement of human DNA: 621, 464 and 157 bp for -/+; 464 and 157 bp for +/+; 621 bp for -/-, respectively.Fig. 1 (top left). All samples in the present study including the nine presented in Fig. 1 (top right) did not contain a XmnI site in the Aγ-globin promoter, thus resulting in 624-bp fragments in all cases. Gγ- and Aγ-globin PCR products were digested with DraI restriction enzyme as a control for the specificity of amplification. Dra I recognition sequence is a single nonpolymorphic site in bothγ-globin promoter fragments, and upon digestion with this restriction enzyme, Gγ- (Fig. 1, bottom left) and Aγ- (Fig. 1, bottom right) globin PCR products yielded 391- and 233-bp DNA fragments in all samples.

Analysis of -158(C → T) (XmnI) polymorphism. XmnI polymorphism in amplified Gγ- andAγ-globin promoter DNA fragments. Top left, XmnI restriction analysis of Gγ-globin promoter. Digestion byXmn I of Gγ-globin promoter DNA fragment (621 bp) results in the appearance of 464- and 157-bp fragments. The presence (+) and absence (-) of XmnI site [-158(C → T mutation)] on one (+/-), or both (+/+ and -/-, respectively) chromosomes of each newborn infant is indicated above. Top right, XmnI restriction ofAγ-globin promoter DNA fragment (624 bp). Middle: DraI restriction analysis of Gγ- (left) andAγ-globin (right). Because DraI is nonpolymorphic site, both promoter fragments yield 391- and 230-bp fragments. Bottom, schematic diagrams of Gγ- (left) andAγ-globin (right) promoter DNA fragments indicating the locations of XmnI (X) and DraI (D) sites and the products of enzymatic digestion.
To validate the analysis of -158(C → T) polymorphism by the PCR-based method, the incidence of XmnI site in the nine samples shown in Fig. 1 was also ascertained by an alternative method, Southern analysis of the genomic DNA. The cellular DNA isolated from cord blood was digested with XmnI, resolved on agarose gels, transferred to nylon membranes by capillary blotting and probed with [32P]-labeledGγ-globin IVS-II DNA fragment(7). All nine samples shown in Fig. 1, as well as 10 additional DNA samples, resulted in hybridization signals in Southern blots in agreement with the results obtained from the PCR-based method [data not shown, was similar to what others have described(7)].
The results obtained from the cord blood of 59 normal term infants where gestational age was carefully monitored are shown in Table 1. The data illustrate that the level HbF synthesis and Gγ globin chain production are greater in term newborns with the XmnI genotype. The actual HbF percentages in the cord blood were not significantly different; however, the ratio of Gγ globin was increased in the presence of the C → T variation.
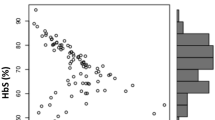
The distribution of total HbF and the Gγ component as well as the percent of synthesis in relation to the presence or absence of theXmn I polymorphism are shown in Fig. 2. The figure shows the dispersion of these values at term gestation and illustrates that the mean values are effected by the genotype. Fetal Hb synthesis, especially the proportion of the Gγ component, is increased in the presence of XmnI polymorphism. The DNA analysis of 100 infants revealed that an incidence of C → T polymorphism in a Caucasian French Canadian population could be extrapolated as 0.32 (+/+:11, +/-:41, and-/-:48). The incidence of polymorphism was obtained by dividing the number ofXmn I-positive chromosomes (63) by the total number of chromosomes analyzed (200).

The percentages of (A) HbF, (B)Gγ, (C) HbF, and (D) Gγ synthesis in relation to the (+/+), (+/-), and (-/-) genotypes. (•) represents the means and (-) SDs. * denotes both a significant analysis of variance and a post test for linear trend.
DISCUSSION
This report shows that a DNA polymorphism of the XmnI site at-158 bp from the cap site of the Gγ gene was found to be associated with delayed switchover from HbF to HbA during the perinatal period. However, at the postconceptional age corresponding to term, no significant effect could be detected on the total HbF concentration; this is similar to what has been reported by others who measured total HbF only in relation to XmnI polymorphism(13).
The lack of significant effect can be explained by the life span of the fetal erythrocytes. Because each erythrocyte remains in the circulation for about 2-3 mo, the proportion of HbF in the oldest erythrocytes in a cord blood sample from a term infant reflects the amounts of HbF that were being produced in an immature fetus (e.g. 28-30 wk of gestation) which is around 90-95%. This high proportion of HbF in the immature erythrocytes entering the circulation changes little as gestational age progresses beyond 32 wk. The HbF in a hemolysate obtained from the cord blood of a term infant originates from red cells having been produced during a period of development ranging from approximately 30-40 wk of gestational age. Although there could be other unknown modifying factors affecting total HbF concentration, it is understandable that the relative proportion of HbF being synthesized by immature red cells in peripheral blood provides a more precise correlation with gestational age than the values of HbF obtained by column chromatography alone(14).
The results of this study show that fetuses with XmnI polymorphism have a slower switchover from HbF to HbA synthesis. The delay in switchover in infants having the C → T variation should manifest itself with higher HbF in the peripheral blood of these infants within the first few months after a term birth. The incidence of -158(C → T) DNA polymorphism in Caucasian French Canadian newborn infants was found to be 0.32 in this study. This frequency of -158(C → T) DNA polymorphism in our study group was higher than those reported in normal Japanese (0.15) and Korean (0.16)(15) and Saudi (0.1)(16) populations but is similar to what has been observed in European populations(13, 17).
The findings of the present study show that DNA sequence variation of C→ T is associated with a higher HbF expression during the switchover from HbF to HbA synthesis. The increased synthesis of HbF is most likely due to the effect of the increased synthesis of the Gγ fraction. TheXmn I polymorphism is in part responsible for the large variation in HbF synthesis found in term infants. This information provides additional knowledge that could be helpful toward understanding the control of globin gene regulation and Hb switching.
Abbreviations
- HbA:
-
adult Hb
- HbF:
-
fetal Hb
- PCR:
-
polymerase chain reaction
References
Stamatoyannopoulos G, Nienhuis A 1994 Hemoglobin switching. In: Stamatoyannopoulos G, Nienhuis A, Majerus P, Varnus H (eds) The Molecular Basis of Blood Diseases. WB Saunders, Philadelphia, pp 107–155
Bard H 1975 The postnatal decline of hemoglobin F synthesis in normal full term infants. J Clin Invest 55: 395–398
Bard H, Widness JA, Ziegler EE, Gagnon C, Peri KG 1995 The proportions of Gγ- and Aγ-globins in the fetal hemoglobin synthesized in preterm and term infants. Pediatr Res 37: 361–364
Wood WG, Bunch C, Kelly S, Gunn Y, Breckon G 1985 Control of haemoglobin switching by a developmental clock?. Nature 313: 320–323
Papayannoupoulou T, Kalmantis TH, Stamatoyannopoulos G 1979 Cellular regulation of hemoglobin switching: evidence for inverse relationship between fetal hemoglobin synthesis and degree of maturity of human erythroid cells. Proc Natl Acad Sci USA 76: 6420–6424
Cunningham JM, Jane SM 1996 Hemoglobin switching and fetal hemoglobin reactivation. Semin Hematol 33: 9–23
Gilman JB, Huisman THJ 1985 DNA sequence variation associated with elevated fetal Gγ-globin production. Blood 66: 783–787
Sampietro M, Thein SL, Contreras M, Pazmany L 1992 Variation of HbF and F-cell number with the G-γ XmnI (C → T) polymorphisms in normal individuals. Blood 79: 832–833
Lolis D, Georgiou I, Loizou P, Makrydimas G 1995 High HbF in pregnancy is associated with the XmnI polymorphism at the -158 bp of the Gγ-globin gene. Eur J Obstet Gynecol Reprod Biol 60: 153–156
Rosatelli MC, Altay C, Oner R, Leoni GB, Moi B, Atzori G, Cao A 1992 β-globin haplotype and XmnI polymorphism at position Gγ-158 and HbF production in Fanconi's anemia. Haematologica 77: 106–109
Shimizu K, Keino H, Terasawa T, Shichishima T, Ikuta K, Hayashi Y 1988 Elevated haemoglobin F in juvenile and adult chronic myelogenous leukaemia. Acta Haematol 80: 28–33
Shen SH, Slighton JL, Smithies O 1981 A history of globin gene duplication. Cell 26: 191–203
Rochette J, Dodé C, Leturcq F, Krishnamoorthy R 1990 Level and composition of fetal hemoglobin expression in normal newborn babies are not dependent on β cluster DNA haplotype. Am J Hematol 34: 223–224
Bard H, Makowski EL, Meschia G, Battaglia FC 1970 The relative rates of synthesis of hemoglobins A and F in immature red cells of newborn infants. Pediatrics 45: 766–772
Shimizu K, Park KS, Enoki Y 1992 The XmnI site 5′ to the Gγ-globin gene polymorphism and its relationship to%HbF and%Gg-in normal Japanese and Korean adults. Hum Hered 42: 253–258
El-Hazmi MAF 1989 XmnI polymorphism in theγ-globin gene region among Saudis. Hum Hered 39: 12–19
Efremov GD, Gjorgovski I, Stojanovski N, Diaz-Chico JC, Harano T, Kutlar F, Huisman THJ 1987 One haplotype is associated with the Swiss type of hereditary persistence of fetal hemoglobin in the Yugoslavian population. Hum Genet 17: 132–136
Author information
Authors and Affiliations
Additional information
Supported by the Medical Research Council of Canada (Grant MA 11552).
Rights and permissions
About this article
Cite this article
Peri, K., Gagnon, J., Gagnon, C. et al. Association of -158(C → T) (XmnI) DNA Polymorphism inGγ-Globin Promoter with Delayed Switchover from Fetal to Adult Hemoglobin Synthesis. Pediatr Res 41, 214–217 (1997). https://doi.org/10.1203/00006450-199702000-00010
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199702000-00010
This article is cited by
-
Effect of Swiss-type heterocellular HPFH from XmnI-Gγ and HBBP1 polymorphisms on HbF, HbE, MCV and MCH levels in Thai HbE carriers
International Journal of Hematology (2014)
-
Influence of Xmn 1Gγ (HBG2 c.-211 C → T) Globin Gene Polymorphism on Phenotype of Thalassemia Patients of North India
Indian Journal of Hematology and Blood Transfusion (2014)
-
The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity
BMC Medical Genetics (2010)
-
The Xmn1 polymorphic site 5′ to the Gγ gene and its correlation to the Gγ:Aγ ratio, age at first blood transfusion and clinical features in β-Thalassemia patients from Western Iran
Molecular Biology Reports (2010)
