
Abstract
Rapid progress in cystic fibrosis research affords the possibility of halting the progress of the lung disease. We used data from 215 patients who had sputum cultures negative for Burkholderia cepacia, at least one outpatient pulmonary function test during 1990, and at least one test a year later to estimate the number of subjects and study duration required to demonstrate that a hypothetical treatment reduces the rate of decline of forced expiratory volume in 1 s (FEV1) to zero. Mean rate of decline of FEV1 (percent predicted) was about 2% predicted per year. Variability decreases with increasing time of observation. For a 1-y study, with α = 0.05 and β = 0.20, over 550 patients must complete the study in each group to show that a treatment halts pulmonary decline. For a 2-y study, 86 subjects in each group are required, and for 4 y, 65. Increasing the number of data points used to determine the rate of decline of FEV1 had only small effect on sample size. Use of pulmonary function data collected at regular intervals for research purposes did not alter these conclusions. Higher initial FEV1 was associated with a greater rate of decline, and among patients with initial FEV1 >60% predicted, younger subjects had a faster decline than did older subjects. Thus, fewer subjects will be required to detect a complete halt in progression of lung disease if the patients are young and have mild pulmonary disease.
Similar content being viewed by others
Main
When CF was first described, patients who did not succumb to meconium ileus in the neonatal period died soon after of staphylococcal pneumonia and/or malnutrition. Treatment of this disease, developed with no specific knowledge of the basic defect, focused on nutritional repletion, vigorous clearance of airway secretions, and aggressive antibiotic therapy(1). This treatment improved the median survival age to 29 y nationwide(2). Now, the discovery of the CF gene and the research this discovery stimulated have brought several treatments directed at correcting or circumventing the basic defect to the clinical arena.
The CF community will soon be faced with the exciting task of testing new treatments that offer the possibility of halting entirely the progress of lung disease, or control of the disease process in the lung at its inception. To test the efficacy of such treatments, outcome measures must be selected that are objective and associated with the ultimate goal-survival. In recent conferences, sponsored by the CF Foundation, the National Institutes of Health, and the Food and Drug Administration, outcome measures for assessing the lung disease of CF were discussed(3). No perfect measure was identified, but pulmonary function tests, particularly the FEV1 and the forced vital capacity, have emerged as important outcome measures to assess lung disease progression. These standardized measures can be obtained repeatedly in patients over the age of 5 y, and the FEV1 correlates with mortality in several studies(4, 5). However, these tests are variable even within a given subject, more so in CF patients than in normal controls(6, 7), and the rate of progression of these markers varies substantially from patient to patient(8).
Some of the treatments aimed at the basic defect in CF are complex and expensive, and entail risk to the patient volunteers. Therefore, it is critical that the early efficacy trials be designed with sufficient power to be definitive. The characteristics of the patient population as well as the duration of the trial will affect our ability to judge the effectiveness of a particular treatment. At our Center, we have a large population of patients who have received aggressive care for decades. These patients have, for the most part, undergone regular pulmonary function testing at times of clinical stability in addition to testing during acute exacerbations, and this information is recorded in a longitudinal relational database. They offer the opportunity to estimate the number of patients and study duration needed to demonstrate efficacy of new therapies directed at the basic defect.
METHODS
Patient population. Patients were selected from the Cleveland CF Center database who had a documented diagnosis of CF by conventional criteria, at least one outpatient pulmonary function test in 1990, and at least one pulmonary function test in the subsequent 4 y more than 1 y from the first. Patients with Burkholderia cepacia in their sputum cultures were excluded. The remaining population consisted of 215 patients. For each of the 215 patients (age 5-40 y) who met these enrollment criteria, subsequent values of FEV1 were selected, where available, at 1-y intervals through 1994. The test closest to 1, 2, 3, or 4 y after the 1990 test was selected for inclusion, which gave up to five measurements per patient for a total of 913 data points.
A second population was patients with CF enrolled in the placebo arm of the 4-y ibuprofen clinical trial, which was previously reported(9). This group was studied to ascertain whether recording of pulmonary function tests at defined intervals under research conditions altered the conclusions from the larger population of patients with CF. This group consisted of 42 patients age 5-39 y with FEV1 > 60% predicted at the beginning of the study period.
Outcome measures. FEV1 was the outcome measure for these studies because, of all the pulmonary functions routinely measured in this Center, the FEV1 best predicts survival. In addition, within-subject reproducibility of the FEV1 and the use of the FEV1 to predict mortality has been reported by others(4, 5). FEV1 was measured with the use of the MedGraphics Pulmonary Function System 1070-1085 (Medical Graphics, St. Paul, MN), according to published standards(10). Percent of predicted normal values obtained from the equations of Knudson et al.(11) were used to allow comparison of results from patients of different height, gender, and age. Because the goal was to follow the change in the FEV1 over time in a patient population, many of whom are growing, use of percent predicted is mandatory, even for within-subject comparisons.
Analysis. Data were analyzed in the mixed model linear regression (SAS Institute Inc, 1993) to estimate the annual rate of decline in FEV1 and its SEM. To do this, annual FEV1 measurements were regressed in time (in years) from the baseline, or 1990, measurement. The results were weighted for each patient according to the number and variability of measurements. Estimated rates of decline for this “control” group (i.e. the actual patient population from our Center) were then compared with a hypothetical treatment group with a mean rate of decline equal to zero and variability equal to the “controls.” The objective of the study was to determine how many subjects need to be studied over what period of time to demonstrate that a therapy directed at the CF basic defect in the lung is curative. We postulated that the treatment being tested halts disease progression, but does not recover pulmonary function which has already been lost, that is, the slope of the treatment group for FEV1 percent predicted versus time is zero. We also assumed that the variability in the FEV1 remains the same as the “control” group. We then determined the rate of decline of percent predicted FEV1, as well as the SD of this measure both in the CF population as a whole and in various subgroups. We used these values to determine, using t test criteria, the number of subjects required to demonstrate a significant difference between the hypothetical treated group (with slope of zero) and a control group with rate of decline as determined from our clinical data, given α= 0.05 and β = 0.2. That is, we determined the number of subjects who would have to be studied in each group to have an 80% probability of demonstrating a true difference between treatment groups at the p< 0.05 level.
We also determined the decline in FEV1 and the variability in this measurement for subjects studied over 1, 2, 3, and 4 y, to determine the effect of study duration on the number of subjects required. We determined, for the 4-y point, whether including more data points between the initial and final points reduces the variability or changed the estimate of the slope and therefore the number of subjects required. We determined the rate of change of FEV1 as a function of initial FEV1.
To test whether the use of pulmonary function values collected for clinical purposes exaggerates the variability (possibly because physicians are more likely to order tests when a patient is worsening or immediately after a successful treatment course to verify success), we also examined results from the placebo-treated patients in the 4-y, double-blind, placebo-controlled trial of ibuprofen in patients with CF(9). In this study, pulmonary function testing was performed at 6-mo intervals during periods of clinical stability. Patients enrolled in this trial were age 5-39 y at the outset and had FEV1 > 60% predicted.
RESULTS
When all patients who met entry criteria are considered, the rate of change of FEV1 was about 2% predicted per year whether it was calculated over a 1-, 2-, 3-, or 4-y period. However, the variability in the rate of change of FEV1 was much greater in the 1-y period than in longer time periods. The variability dramatically affected the sample size needed to detect the effect of a treatment that halts disease progression completely. Increasing study duration resulted in reduced number of subjects required, with the biggest drop occurring between 1 and 2 y duration (Table 1).
We then used the 4-y analysis to determine whether increasing the number of data points collected in the course of the study changed either the calculated slope or the variability. Increasing the number of data points considered in the 4-y analysis had minimal impact on either of these measures, and had only modest impact on the number of subjects required to detect the hypothetical treatment effect (Table 2). For subsequent analyses, therefore, a 4-y study duration and only the beginning and ending points were used.
Further analysis tested whether variables like age or initial pulmonary function affected either the rate of change of FEV1 or the variability. Initial FEV1 affected the rate of decline, expressed as percent predicted: the higher the initial FEV1, the greater the rate of decline over 4 y. The correlation between slope and initial FEV1 was -0.21(p = 0.0006). SD did not vary significantly with initial FEV1. Slope and variability for patients with different initial FEV1 values is shown in Table 3. This table does not include patients enrolled in the ibuprofen arm of the ibuprofen clinical trial, because this drug alters the rate of decline of FEV1, and some of these patients stopped taking the study drug during the period of evaluation. Thus, the variability in their rate of decline may be artificially increased. Fewer subjects are needed to demonstrate the hypothetical treatment effect among patients with normal initial FEV1 than among the sicker patients.
To test whether the large standard deviations observed in short-term follow-up were an artifact of using data collected for clinical purposes, where physicians are more likely to obtain tests when a patient is ill or immediately after a course of treatment when the patient is at his best, we studied the rate of decline and variability in patients enrolled in the placebo arm of the 4-y ibuprofen clinical trial recently completed at our institution(9). Pulmonary function tests were obtained at 6-mo intervals at times of clinical stability. Although the SD was slightly less than was observed in the general population, variability decreased with duration of follow-up (Table 4). Comparison of Table 4 with Table 1 also shows that these research subjects, selected for their good to excellent pulmonary function (FEV1 > 60% predicted) had more rapid decline in FEV1 percent predicted than the population as a whole, as suggested by the results in Table 3. Because of this higher rate of decline and slightly lower standard deviations, the number of subjects required to determine that a treatment halts disease progression is much less for the ibuprofen study population than for a study population selected from the general CF population (compare Table 1 with Table 4).
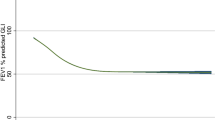
To assess the effect of age on the rate of decline of FEV1, and to avoid the confounding effect of baseline FEV1, we used the patients enrolled in the placebo arm of the ibuprofen study, whose initial FEV1 was at least 60% predicted. Annual rate of decline of FEV1 correlated significantly with patient age at the beginning of the study: older patients had slower rate of decline than younger patients (Fig. 1).

Rate of change of FEV1 as a function of age in patients with CF who were enrolled in the placebo arm of the ibuprofen clinical trial. At the outset, all patients had FEV1 > 60% predicted.
DISCUSSION
New strategies for treatment of CF lung disease are being developed which should correct or circumvent the basic defect. Gene therapy is already in clinical trial(12–14). The use of amiloride to block the excessive sodium reabsorption in combination with uridine triphosphate or its analogs to activate an “alternative” apical chloride channel might approximate normal handling of chloride and sodium ions in the CF airway(15). Other treatments, such as strategies to activate mutant forms of the CF gene product, are in the preclinical phase(16–18). If these new therapies directed at the basic defect are to be accepted as“cures,” they must be shown to prevent the fatal lung disease. Such a demonstration is complicated, however, by the improved prognosis of CF with only symptomatic treatment. The median survival age is now nearly 30 y(2), and the rate of deterioration of lung function is slow and variable. To distinguish the current very slow deterioration from no deterioration at all, large numbers of patients may need to be studied over long periods of time.
The number of patients needed to detect a clinically important difference between treated and control groups depends on the magnitude of the expected difference between the groups and the variability of the measure. Our results suggest that the variability of the slope of FEV1 with time decreases as the duration of the study increases, so detecting a curative therapy in a study period of 1 y or less will require a very large number of study subjects.
At first glance, it may seem best to conduct a trial of an unproven therapy directed at cure on patients old enough to consent for themselves, and already with moderate disease, so that, if a disastrous result occurs, fewer years of life and health are lost. However, our data suggest that testing curative therapies, which may be expensive and/or cumbersome to deliver, will be done most efficiently in young patients with excellent pulmonary function. There are, in addition, biologic reasons to consider testing in young, healthy CF patients. CF patients with established bronchiectasis may not benefit from interventions directed at the cause of the disease. Indeed, postinfectious bronchiectasis often is progressive despite removal of the initiating stimulus(19). Thus, treatments directed at the basic defect may be most effective in patients with normal or near-normal lungs. Moreover, this group has the most to gain in terms of years of useful and symptom-free life.
Our assumptions clearly affect our results. We chose to follow the rate of change of the FEV1 over time, rather than a short-term change in FEV1, because improving the FEV1 acutely without altering the long-term course does not represent a cure. The choice of the FEV1 as an outcome measure was based on our conviction that a cure must affect mortality, and a clinical measure that can serve as a surrogate marker for mortality must be used in trials seeking a cure. In addition, we assumed that a curative treatment will prevent disease progression but not reverse established disease. If the treatment only slows disease progression, more subjects will be required to demonstrate that. If the treatment reverses disease, then fewer subjects are necessary. The ability to generalize our results depends on how representative the patients at our Center are of the CF population at large. Median survival age for the Cleveland population(calculated over 10 y to get enough deaths for statistical validity) is comparable to the national median survival age reported by the CF Foundation's Data Registry(2). Only a few studies report longitudinal rates of FEV1 decline that can be directly compared with ours. A study on the CF population in Toronto compared the longitudinal rate of FEV1 decline in patients infected with and not infected with Pseudomonas aeruginosa. Patients with P. aeruginosa declined at a rate of 3.5% predicted per year, and those who were not colonized, at a rate of 1.9% predicted per year(20). In the Netherlands, longitudinal data were collected on CF patients during the years that infants were screened for CF and in the subsequent period in which they were not. Patients identified by screening declined at a rate of 1.2% predicted per year when they were old enough to perform pulmonary function tests, and patients diagnosed after the screening program terminated declined at a rate of 2.6% predicted per year(21). All these values are comparable to our values of 2.3% predicted per year. Cross-sectional data from the Netherlands(22) are also concordant. Analysis of data from the Toronto population showed that, although the study group as a whole declined at a rate of 1.25% predicted per year, patients age 6-11 y declined much faster, at a rate of 4.04% predicted per year(23), in keeping with our results. Thus it is likely that the patients in this study are representative of the current status of CF.
Cure of the CF lung disease will still leave the patient with substantial morbidity in other organs. Most CF patients will still have pancreatic insufficiency, some will have pancreatitis. Some will have cirrhosis and die of it. Some will develop diabetes. Intestinal obstruction will persist, and reproductive failure in men will not be improved. But by far the most troublesome clinical problems now arise in the lung, and correction of the basic defect in the lung at a sufficiently early stage in the disease should effectively prevent all pulmonary disease and associated complications.
The rapid pace in CF research should bring us soon to clinical testing of treatments designed to prevent all secondary consequences of the lung disease. The results presented here suggest that our success in managing the lung disease of CF will make meticulous, long-term clinical studies on a particularly vulnerable population necessary to recognize even a treatment that halts the progress of lung disease entirely. Use of outcome measures not clearly associated with mortality, or shorter term studies that focus only on absolute values for pulmonary function and not on the rate of decline will not be satisfactory. Curative therapy must be nontoxic as well as effective to provide a significant advance over conventional therapy in CF.
Abbreviations
- CF:
-
cystic fibrosis
- FEV1:
-
forced expiratory volume in 1 s
References
Davis PB 1994 Evolution of therapy for cystic fibrosis. N Engl J Med 331: 672–673
Fitzsimmons SC 1993 The changing epidemiology of cystic fibrosis. J Pediatr 122: 1–9
Ramsey BW, Boat TF 1994 Outcome measures for clinical trials in cystic fibrosis. J Pediatr 124: 177–192
Kerem E, Reisman J, Corey M, Canny GJ, Levison H 1992 Prediction of mortality in patients with cystic fibrosis. N Engl J Med 326: 1187–1191
Wagener JS, Taussig LM, Burrows B, Hernried L, Boat T 1980 Comparison of lung function and survival patterns between cystic fibrosis and emphysema or chronic bronchitis patients. In: Sturgess JM (ed) Perspectives in Cystic Fibrosis: Proceedings of the Eighth International Cystic Fibrosis Congress, Toronto, May 26-30, 1980. Canadian Cystic Fibrosis Foundation, Toronto, pp 236–245
Cooper PJ, Robertson CF, Hudson IL, Phelan PD 1990 Variability of pulmonary function tests in cystic fibrosis. Pediatr Pulmonol 8: 16–22
Nickerson BG, Lemen RJ, Gerdes CB, Wegmann MJ, Robertson G 1980 Within-subject variability and per cent change for significance of spirometry in normal subjects and in patients with cystic fibrosis. Am Rev Respir Dis 122: 859–866
Corey M, Levison H, Crozier D 1976 Five to seven-year course in pulmonary function in cystic fibrosis. Am Rev Respir Dis 114: 1085–1092
Konstan MW, Byard PJ, Hoppel CL, Davis PB 1995 Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med 332: 848–854
Standardization of spirometry-1987 update: statement of the American Thoracic Society 1987 Am Rev Respir Dis 136: 1258–1298
Knudson RJ, Lebowitz MD, Holbert CL, Burrows B 1983 Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis 127: 725–734
Knowles MR, Hohneker KW, Zhou ZQ, Olsen JC, Noah TL, Hu PC, Leigh MW, Engelhardt JF, Edwards LJ, Jones KR, Grossman M, Wilson JM, Johnson LG, Boucher RC 1995 A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med 333: 823–831
Crystal RG, McElvaney NG, Rosenfeld MA, Chu C-S, Mastrangeli A, Hay JG, Brody SL, Jaffe HA, Eissa NT, Danel C 1994 Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet 8: 42–51
Hay JG, McElvaney NG, Herena J, Crystal R 1995 Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum Gene Ther 6: 1487–1496
Knowles MR, Clarke LL, Boucher RC 1991 Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N Engl J Med 325: 533–538
Eidelman O, Guay Broder C, van Galen PJ, Jacobson KA, Fox C, Turner RJ, Cabantchik ZI, Pollard HB 1992 A1 adenosine-receptor antagonists activate chloride efflux from cystic fibrosis cells. Proc Natl Acad Sci USA 89: 5562–5566
Gribkoff VK, Champigny G, Barbry P, Dworetzky SI, Meanwell NA, Lazdunski M 1994 The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J Biol Chem 269: 10983–10986
Kelley TJ, Al-Nakkash L, Drumm ML 1995 CFTR-mediated chloride permeability is regulated by type III phosphodiesterases in airway epithelial cells. Am J Respir Cell Mol Biol 13: 657–664
Ellis DA, Thornley PE, Wightman AJ, Walker M, Chalmers J, Crofton JW 1981 Present outlook in bronchiectasis: clinical and social study and review of factors influencing prognosis. Thorax 36: 659–664
Kerem E, Corey M, Gold R, Levison H 1990 Pulmonary function and clinical course in patients with cystic fibrosis after pulmonary colonization with Pseudomonas aeruginosa. J Pediatr 116: 714–719
Dankert-Roelse JE, te Meerman GJ 1995 Long term prognosis of patients with cystic fibrosis in relation to early detection by neonatal screening and treatment in a cystic fibrosis centre. Thorax 50: 712–718
Gan K-H, Veeze HJ, Van den Ouweland AMW, Halley DJJ, Scheffer H, VanderHout A, Overbeek SE, DeJongste JC, Bakker W, Heijerman HGM 1995 A cystic fibrosis mutation associated with mild lung disease. N Engl J Med 333: 95–99
Kovesi T, Corey M, Levison H 1993 Passive smoking and lung function in cystic fibrosis. Am Rev Respir Dis 148: 1266–1271
Author information
Authors and Affiliations
Additional information
Supported by National Institutes of Health Grant P30 DK27651 and by a Research Development Program Grant from the Cystic Fibrosis Foundation.
Rights and permissions
About this article
Cite this article
Davis, P., Byard, P. & Konstan, M. Identifying Treatments That Halt Progression of Pulmonary Disease in Cystic Fibrosis. Pediatr Res 41, 161–165 (1997). https://doi.org/10.1203/00006450-199702000-00001
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199702000-00001
This article is cited by
-
Gene therapy for cystic fibrosis: an example for lung gene therapy
Gene Therapy (2004)
-
Macrolides in Cystic Fibrosis
American Journal of Respiratory Medicine (2002)
