
Abstract
Seventy-eight prepubertal, non-GH-deficient children aged 8.1 ± 0.2 y, with very short stature (mean, -3.2 SD) of intrauterine onset, were treated for 3 y with GH [0.4 (dose D1) or 1.2 (dose D2) IU/kg/wk] and 66 were followed during a 4th y without GH therapy. A 2-y intermediary report had demonstrated a GH dose-dependent acceleration of growth. During the 3rd y on GH, patients D2 (1.2 IU/kg/wk) continued with the same dose, whereas patients D1 (0.4 IU/kg/wk) were randomized to either continue on D1 (group D1) or be increased to D2 (group D1D2). After 3 y on GH, patients' mean height (SD) reached -2.37(D1), -2.17 (D1D2), and -1.58 (D2) with a total mean height gain of 0.77 (D1), 0.93 (D1D2) (difference NS), and 1.61 SD (D2 significantly higher than D1 and D1D2, p ≤ 0.0001). During the off-treatment year, mean growth rate (cm/y) decreased to 3.4 in patients D1, 3.7 in D1D2, and 4.1 in D2 (NS). During the 4 y, bone age advanced of 4.6, 4.6, and 5.3 y in D1, D1D2, and D2, respectively, and puberty started in 34 patients (10 during the off-treatment year). Age at onset of puberty, apparently within normal range, did not relate either to the dose or the duration of treatment. Clinical and biologic tolerance of treatment was good. In conclusion this study demonstrates a GH dose-dependent effect on growth acceleration in persistent postnatal severe growth retardation of intrauterine onset. This effect was sustained for 3 y at 1.2 IU/kg/wk followed by a peculiar growth deceleration at treatment discontinuation. Additional studies are necessary to optimize long-term GH treatment regimen and to document its effects on final height.
Similar content being viewed by others

Main
IUGR is defined as birth weight and/or length below -2 SD for gestational age. These conditions are encountered in approximately 2.5% of newborns. Postnatal catch-up growth usually occurs spontaneously before age 4 y, in at least 80% of these children(1). Those whose height remains subnormal after 4 y are defined as IUGR short children.
Short-term results of treatment with GH in such short children born with IUGR have been reported by various authors(1–7), including our group(8). Very few results after 3 y are documented(9). The purpose of this study is to present both the effects of GH treatment given for 3 y to very short prepubertal children with IUGR and the outcome of growth during a 4th y without treatment.
METHODS
The trial started in February 1988. The first 6-mo period was designed as a randomized, double-blind, placebo-controlled study with two doses (0.4 and 1.2 IU/kg/wk) of GH (Maxomat®, Sanofi Winthrop, France) in severely growth-retarded prepubertal IUGR children from parents of normal height. The protocol was approved by the Comité d'Ethique des Hôpitaux de Paris. After individual written informed consents were obtained, 92 IUGR patients were included. Details regarding patients, methods, and results in this first phase of the study have been recently published(8). A significant dose-dependent growth acceleration during the first 2 y on GH was demonstrated. Ethically approved amendments added a 3rd y on GH and 1 y of posttreatment study.
Age at admission in the trial was 4-11 y in boys and 4-10 y in girls. Both birth length and birth weight had to be -2 SD or more below the mean for gestational age, according to Usher and Mac Lean(10). Height at admission had to be -2 SD or more below the mean according to the usual French standards(11), and the weight within±2SD for height, the bone age being either retarded or at most equal to age. Annual growth velocity had to be documented for at least the last 12 mo, and not to exceed the normal mean for age according to the same standards(11). Height of both parents had to be within normal limits for French standards(11). The patients had to be strictly prepubertal according to the whole criteria of Marshall and Tanner(12, 13). Serum GH response had to be at least 10 ng/mL at one conventional stimulation test or at least 7.5 ng/mL at two different tests. Criteria for exclusion of the trial were: children born from multiple pregnancies, or with gestational age below 34 wk; children with known embryo fetopathy or with malformations; children with chromosomal abnormality or with dysmorphic syndromes other than that of Silver-Russell (only six Silver-Russel patients were included); children with any chronic illness; children with low serum thyroxine or with detectable abnormality of glycemic control; children who had received any previous hormonal treatment.
After 2 y of GH treatment, ethically approved amendments and individual informed consents were obtained to continue GH therapy for a 3rd y and to realize a follow-up study without GH treatment for a 4th y. Randomization at the onset of the study had created two groups of children receiving GH daily, 6 d a week at bedtime, as s.c. injections of either 0.4 IU/kg/wk (D1 dose) or 1.2 IU/kg/wk (D2 dose), as detailed in the 2-y report(8). At the end of the 2nd y, the patients who had completed 2 y of treatment at a dosage of 0.4 IU/kg/wk were randomized again to receive, during the 3rd y, either the same dosage (group D1) or a dose of GH switched to 1.2 IU/kg/wk (group D1D2). For the patients who had entered the study with GH 1.2 IU/kg/wk (group D2), no change in dose other than a regular adaptation to weight was introduced.
Clinical follow-up consisted of visits every 3 mo during GH treatment and every 6 mo during the posttreatment year, in 10 centers in France and Belgium participating in the study(8). The height recorded at each visit was the mean of three successive measurements performed by the same pediatrician or pediatric nurse using the same wall-mounted stadiometer. Sexual development was evaluated on testicular volume in boys and breast volume in girls, and assigned to pubertal stages 1-5 according to Marshall and Tanner(12, 13). X-ray of the hand and wrist was done every 6 mo for evaluation of bone age according to Greulich and Pyle(14). Bone age was further centrally rated by two independent and skilled pediatric radiologists working in a blind fashion, and the mean of their ratings was used. Blood samples were collected every 6 mo for local routine examinations, central measurement of serum IGF-I, and central assay of anti-GH and anti-ECP antibodies. Fifty patients underwent an oral glucose load test between the 18th and 24th mo. The methods have been published in detail elsewhere(8).
Comparability of patients' auxologic main characteristics between the different dose groups at randomizations were tested using analysis of variance or the Fisher exact test. Statistical tests were two-sided, with an α level of 0.05. Results are expressed as mean ± SEM values.
RESULTS
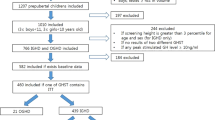
Table 1 summarizes the pertinent pretreatment clinical characteristics of the 78 IUGR children who had completed 3 y of GH therapy.Figure 1 shows the number of patients at each stage of the study. At the onset of the 3rd y on GH, group D1 consisted of 15 patients(10 male, 5 female), group D1D2 of 25 patients (16 male, 9 female), and group D2 of 44 patients (26 male, 18 female). During the 3rd y, six children dropped out of the trial for weariness of injections and, during the 4th y, 12 for noncompliance to the protocol. At the end of the 4th y, 66 patients (11 D1, 21 D1D2, and 34 D2) were still in follow-up.

Schematic representation of the number of patients.
Mean annual height velocities, expressed in centimeters/y, are shown inTable 2, and in SD in Figure 2. In group D1, height velocity was successively 1.48 ± 0.50, 0.56 ± 0.39, and 0.05 ± 0.30 SD during the 1st, 2nd, and 3rd y on GH treatment and -1.25 ± 0.15 SD during the posttreatment year of follow-up. In group D1D2, the mean height velocity of the first 2 y (1.50 ± 0.32 and 0.45 ± 0.39 SD) was close to that of D1; on the 3rd y, with D2 dose, it increased to 1.06 ± 0.43 SD, significantly greater than the D1 group value (p = 0.02); during the posttreatment year, it dropped to -1.73± 0.39 SDS. In group D2, mean height velocity sharply increased to 3.77± 0.31 SD in the 1st y, then decreased to 1.64 ± 0.25 and 1.08± 0.27 SD in the 2nd and the 3rd y, this growth rate being significantly higher than that of the other groups during the first 2 y. In the 3rd y, growth rate in group D2 was not significantly different from that of group D1D2; in the 4th y, without GH, growth rate in patients D2 dropped to-1.32 ± 0.24 SD.

Mean (±SEM) annual height velocity, expressed as SD for age, at annual visits.
A waning effect of GH, at unchanged dose/kg, was observed during the 2nd and 3rd y in patients D1 and D2, similar to that described in patients treated for hypopituitarism or Turner syndrome. Tripling the GH dose after 2 y in patients D1D2 resulted in a definite but nonsignificant increase of growth velocity. However, as shown in Figure 2, giving 1.2 IU/kg/wk to these patients during the 3rd was far from producing the same sharp acceleration of growth as in the 1st y (p < 0.001).
There was a wide range of individual variations. Thirty-one patients had an annual growth rate of at least 1 SD above the normal mean during the 3rd y: 2/15 (13.3%) in group D1, 10/23 (43.4%) in group D1D2, and 19/40 (47.5%) in group D2.
At the end of 3 y of GH treatment, height (mean ± SEM) reached -2.37± 0.28 SD for age in patients of group D1, -2.17 ± 0.24 in group D1D2, and -1.58 ± 0.14 SD in group D2 (Fig. 3). The total height gain was 0.77 ± 0.10 SD in group D1, 0.93 ± 0.15 in group D1D2, and 1.61 ± 0.08 in group D2, significantly higher in D2 patients than in those of the other two groups (p < 0.001). The percentage of children whose height was then within the normal range was 46.7% (7/15) in group D1, 52.2% (12/23) in group D1D2, and 70% (28/40) in group D2, respectively. Statistical analysis within group D1 showed that height gain had been 0.96 ± 0.12 SD in eight patients whose initial height was above -3 SD, significantly better than in seven other patients whose initial height was below -3 SD and who gained only 0.55 ± 0.12 SD(p < 0.004).

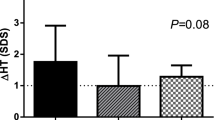
Mean (±SEM) height, expressed as SD for age at annual visits. The data are generated from the same patients over 4 y (D1,n = 11; D1D2, n = 17; D2, n = 29) followed longitudinally.
During the year of follow-up without treatment, a decelerated growth was observed in most patients: mean growth velocity fell below -1 SD(Table 2 and Fig. 2). It was not significantly different among the three groups. The pattern of growth during this year off GH treatment seemed biphasic, a more important deceleration being observed during the first 6 mo than during the next semester.
As shown in Figure 3, this catch-down resulted in a mean loss in height of approximately 0.25 SD for age. At the end of the 4th y of the study, the mean height was in the normal range for group D2 (-1.76± 0.17 SDS); this was better than in groups D1 (-2.46 ± 0.39 SD) and D1D2 (-2.41 ± 0.29 SD), the difference being close to significance with D1 (p = 0.06) and significant with D1D2 (p = 0.04). In spite of growth deceleration after the end of treatment, the total height gain over 4 y of follow-up was 1.32 ± 0.13 SD in D2 versus 0.55 ± 0.15 in D1 (p < 0.004) and 0.70 ± 0.17 in D1D2 (p < 0.007), thus significantly better in patients treated with 1.2 IU/kg/wk for 3 y than in those who started with 0.4 IU/kg/wk, irrespective of the increased dose for the 3rd y.
Weight at the end of the follow-up period, expressed as SD for height(Tables 3 and 4), had increased from the onset of the study but remained in the normal range, slightly above the average.
As shown in Table 4, skeletal maturation (mean± SEM) over 36 mo of GH treatment was, respectively, 41.3, 40.7, and 45.7 mo in groups D1 (n = 11), D1D2 (n = 21), and D2(n = 37). The difference between groups is not significant. The difference with the time elapsed is significant in group D2 (p < 0.001), not in groups D1 and D1D2. During the 4th y, mean skeletal maturation over 12 mo was 14.7 ± 3.8 mo in group D1 (n = 7), 14.8± 1.9 in group D1D2 (n = 12), and 18.4 ± 1.7 mo in group D2 (n = 19). Thus, as a whole for the 4 y of the study, the progression of bone age had been of 4.6 ± 0.7 y in group D1, at low GH dose, identical (4.6 ± 0.4 y) in group D1D2, and not significantly different from the time elapsed. A higher progression was observed in group D2, whose bone maturation reached 5.3 ± 0.4 y in 4 y of follow-up; this value being significantly different from the time elapsed (p < 0.001) but not from the progression values observed in groups D1 and D1D2. However, at the end of the 4th y of study, mean bone age was still retarded in the three groups: at a mean chronologic age of 12.5, 11.9, and 11.9 y, bone age was 10.7, 11.0, and 11.0 in groups D1, D1D2, and D2, respectively(Table 3).
Puberty started in 34 patients during the 3 y on GH, and in 10 others during the 4th y off GH. Among these 44, there were 23 boys reaching a testicular volume ≥ 4 mL at a mean age of 12.6 y with a mean bone age of 11.3 y, and 21 girls whose breast development (Tanner's stage B2) started at a mean age of 11.0 y with a mean bone age of 9.7 y. There were no significant differences either in the frequency of occurrence of puberty or in age at its onset between the three groups. Moreover, age at onset of puberty was not related to the time elapsed from the onset of GH treatment, which varied from 1 to 3 y. The rate of sexual maturation after its onset did not seem to be different between groups or different from the average(12, 13). The possible effects of puberty on growth velocity were analyzed. In each of the three groups and for each year of the study, the annual growth rate of children who remained at pubertal stage 1 did not differ from that of the whole group.
Annual results of the longitudinal follow-up of serum IGF-I, performed with the same RIA in the same laboratory, are shown in Table 5. The changes observed during the first 2 y on GH have been previously reported(8). During the 3rd y, IGF-I levels increased again in all groups; this increase being significant in D2 and close to significance in D1D2, whose dose of GH was tripled (p = 0.087).
Clinical tolerance of treatment was good. The events noted during the first 2 y on GH were detailed in our first report(8). The only adverse events reported later were one isolated seizure during the 3rd y on GH in one child, and one in another patient during the posttreatment year. These seizures were brief, did not repeat, and were not followed by any detectable neurologic or electroencephalographic abnormality.
Controls of routine biologic parameters, including serum thyroxine, did not show any significant change during the study. No significant changes of fasting glycemia and glycosylated Hb were found at any time of the study, although a few children had transient elevations of one of these parameters. At the end of the 3rd y on GH, all patients had normal glycemia and HbA1C, without difference between groups. Oral glucose load, performed in 50 children at the end of the 2nd y of GH treatment, showed a mean glycemia peak of 8.0± 0.3 mmol/L and a mean insulin peak of 42.5 ± 3.4 pmol/L, without a significant difference between patients treated with either 0.4 or 1.2 IU/kg/wk of GH.
During the 3 y on GH no patient had detectable anti-ECP antibodies. Anti-GH antibodies were detected at a level of 5% in 2 of 78 children, without change in their growth velocity.
DISCUSSION
In the initial report of this study(8), the short-term effectiveness of GH and the dose-dependence of its growth-accelerating effect in prepubertal children with very short stature of intrauterine onset were demonstrated. This first report also discussed a possible acceleration of bone maturation and questioned the possibility to maintain the improved position of these children on the growth chart after discontinuation of GH therapy.
The data presented here confirm that GH treatment at sufficient dose continues to accelerate the growth of IUGR short children beyond the 2nd y of treatment, in spite of the well known “waning effect” of GH through the years. They also demonstrate that the growth acceleration induced by a same dose of 1.2 IU/kg/wk is greater when given immediately from the start than after 2 y. This fact is comparable to that observed in IUGR by others(9) whose patients showed a dose-dependent growth velocity in the 1st y on GH, but did not accelerate their growth when the dose was doubled in the 2nd y. The cost-benefit ratio of a high GH dose seems better at onset than when delivered secondarily.
These data and those reported in IUGR short children by other studies(1–9) allow us to conclude that GH treatment of these children induced a catch-up growth for at least 2 y, and for 3 y if the dose is sufficient, and has not shown, up to now, any unexpected or undesirable side effect on the health of the patients. Although mean IGF-I values doubled during the first 2 y of GH treatment and still increased during the 3rd y, they stayed within normal range for age, as shown by their expression in SD (Table 5). No correlation was found between IGF-I levels and growth velocity in the patients of this study.
However, the present data, as those of Albanese and Stanhope(9), show some degree of acceleration of bone maturation in patients treated 3 y with the highest doses of GH. It remains to be seen what benefit in the adult height SD improvement will be observed. Adult height prediction in IUGR is extremely difficult, as previous reports(15–17) have pointed out in such patients a spontaneous acceleration of bone maturation in late childhood and adolescence without the corresponding growth benefit.
In this homogeneous group of patients, some peculiar aspects deserve special attention. The first one is the homogeneity of the cohort, excluding familial short stature, and limiting admission in the trial to children of small birth length at term of at least 34 wk of a single pregnancy, with birth weight corresponding to the length, and exclusion of malformative or dysmorphic syndromes except six cases of Silver-Russell type. A second one is that puberty started in these patients at a mean age that seems within the range of normal children, as previously reported in untreated IUGR children(15–17); mean bone age in girls at the start of puberty could be relatively low (9.7 y), but the number of patients is too small, and actualized normative data are lacking to draw any conclusions. The third is the description of growth velocity after discontinuation of GH treatment. It results in loss of the benefit observed during the 3rd y of treatment. The biphasic pattern of growth rate during the posttreatment year could suggest that this “catch-down” phenomenon could be limited in time.
The fact that annual growth rate during treatment with GH did not differ, in prepubertal patients, from that of the whole group, suggests a poor pubertal acceleration of growth in GH-treated IUGR patients. This is possibly related to the usual poor pubertal growth of untreated IUGR patients(15–17).
The increase in levels of serum IGF-I during the 3rd y of treatment with GH can be possibly related to the onset and/or progress of puberty in 34 of the 78 patients. This increase in IGF-I contrasts with continuous deceleration of growth velocity. This could lead to speculate for low sensitivity to IGF-I in some IUGR short children, apart from others that could be partially GH-deficient(18).
The question of the final height remains unsolved for the patients of this cohort as for those of other studies using GH in endocrinologically normal short children with a history of IUGR. Adult height of the patients will have to be compared with the mean height of untreated individuals with a similar history, which has been reported to be below -2 SD(19). The importance of IUGR as a cause of subnormal adult stature has been recently estimated by follow-up of a large cohort of full-term singleton individuals from birth up to age 18 y(20): 7.9% of those born with subnormal length below -2 SD were found to have a final height in the same range, and 22% of the total short adult population consisted of individuals who had been IUGR newborns.
In conclusion, our data show that treatment with GH is beneficial for longitudinal growth in IUGR short children. However, several important questions remain unanswered. They require additional studies or new therapeutic trials: at what dose, when, and for how long should GH be delivered? The 3-y data agree with the possibility of some loss of GH efficacy with time as hypothesized in the 2-y report(8). Another question is how to determine whether, in growth retardation of intrauterine onset, continuing a high GH dose up to an age close to the end of statural growth would be of real benefit for the patients. A corollary to this issue is raised of what should be done, once the catch-up has been obtained: intermittent treatment? low GH dose for maintenance? discontinuation of treatment? Further long-term clinical research is required to solve the many remaining issues.
Abbreviations
- IUGR:
-
intrauterine growth retardation
- ECP:
-
Escherichia coli protein
References
Albertsson-Wikland K 1989 Growth hormone secretion and growth hormone treatment in children with intrauterine growth retardation. Acta Paediatr Scand(suppl 349: 35–41
Lanes R, Plotnick LP, Lee PA 1979 Sustained effect of human growth hormone therapy in children with intra-uterine growth retardation. Pediatrics 63: 731–735
Rochiccioli P, Tauber M, Moison P, Pienkowski C 1989 Investigation of GH secretion in patients with IUGR. Acta Paediatr Scand(suppl 349: 42–46
Stanhope R, Preece MA, Hamill G 1991 Does growth hormone treatment improve final height attainment of children with intrauterine growth retardation?. Arch Dis Child 66: 1180–1183
Chatelain P on behalf of the International Board of KIGS 1993 Auxology and response to GH treatment of patients with IUGR or Silver-Russell syndrome: analysis of data from the KIGS. Acta Paediatr Scand( suppl 391): 79–81
Czernichow P, Rappaport R 1994 Growth Hormone and Somatomedin during Life-span, Springer, Berlin, pp 161–170
Chaussain JL, Colle M, Landier F 1994 Effects of growth hormone therapy: prepubertal children with short stature secondary to intrauterine growth retardation. Acta Paediatr Scand(suppl 399: 74–75
Chatelain P, Job JC, Blanchard J, Ducret JP, Olivier M, Sagnard L, Vanderschueren-Lodeweyckx on behalf of the Belgian and French Pediatric Clinics and Sanofi 1994 Dose-dependent catch-up growth after 2 years of growth hormone treatment in intrauterine growth-retarded children. J Clin Endocrinol Metabol 78: 1454–1460
Albanese A, Stanhope R 1993 Growth and metabolic data following growth hormone treatment of children with intrauterine growth retardation. Horm Res 39: 8–12
Usher R, Mc Lean F 1969 Intrauterine growth of live-born Caucasian infants at sea level: standards obtained from measurements in 7 dimensions of infants born between 25 and 44 wk of gestation. J Pediatr 74: 901–910
Sempe M, Pedron G, Roy MP 1979 Auxologie, Méthodes et séquences. Theraplix publ., Paris
Marshall WA, Tanner JM 1969 Variations in the pattern of pubertal changes in girls. Arch Dis Child 44: 291–303
Marshall WA, Tanner JM 1970 Variations in the pattern of pubertal changes in boys. Arch Dis Child 45: 13–23
Greulich WW, Pyle SI 1959 Radiographic Atlas of Skeletal Development of the Hand and Wrist, 2nd Ed. Stanford University Press, Stanford, CA
Tanner JM, Lejarriaga H, Cameron N 1975 The natural history of the Silver-Russell syndrome: a longitudinal study of 39 cases. Pediatr Res 9: 611–623
Job JC, Rolland A 1986 Histoire naturelle des retards de croissance a debut intrauterin. Arch Fr Pediatr 43: 301–306
Davies PSW, Valley R, Preece MA 1988 Adolescent growth and pubertal progression in the Silver-Russell syndrome. Arch Dis Child 63: 130–135
DeWaal WJ, HokkenKoelega ACS, Stijnen T, KeizerSchrama SMPFD, Drop SLS 1994 Endogenous and stimulated GH secretion, urinary GH excretion, and plasma IGF-I and IGF-II levels in prepubertal children with short stature after intra-uterine growth retardation. Clin Endocrinol 41: 621–630
Chaussain JL, Colle M, Ducret JP 1994 Adult height in children with prepubertal short stature secondary to intrauterine growth retardation. Acta Paediatr Scand(suppl 399: 72–73
Karlberg J, Albertsson-Wikland K 1995 Growth in full-term small-for-gestational-age infants: from birth to final height. Pediatr Res 38: 733–739
Author information
Authors and Affiliations
Additional information
Participating pediatric clinics in Belgium: Antwerpen (M. DuCaju), Brussels(C. Heinrich, G. Van Vliet), Gent (M. Craen), Liège (L. Thiry), and in France: Bordeaux (M. Colle), Paris (F. Girard, J. E. Toublanc), Rennes (M. Lecornu), Strasbourg (J. G. Juif), Tours (F. Despert). Participating persons in the Sanofi-Choay staff in France: J. Blanchard, P. Carita, S. Claudel, E. Masuy, and L. Sagnard.
Rights and permissions
About this article
Cite this article
Job, J., Chaussain, J., Job, B. et al. Follow-Up of Three Years of Treatment with Growth Hormone and of One Post-Treatment Year, in Children with Severe Growth Retardation of Intrauterine Onset. Pediatr Res 39, 354–359 (1996). https://doi.org/10.1203/00006450-199602000-00027
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199602000-00027
This article is cited by
-
Growth hormone treatment for short stature in children born small for gestational age
Advances in Therapy (2008)
