
Abstract
Near-infrared spectroscopy is a noninvasive technique which measures oxidized cytochrome aa3, oxygenated Hb, and deoxygenated Hb and calculates total Hb in tissue. This technique, in conjuction with measurement of cerebral blood flow, was used in rabbits with experimental bacterial meningitis to determine whether there was evidence for cerebral energy depletion and alterations in the cerebral vascular bed with infection. Rabbits with meningitis had a significant reduction in cerebral blood flow, cerebral oxidized cytochrome aa3 and a relative increase in the deoxygenated Hb fraction and a decrease in the oxygenated Hb fraction compared with uninfected controls. Total Hb was not significantly different between the two groups. These findings may help clarify the mechanism for some of the intracranial pathophysiologic abnormalities in meningitis.
Similar content being viewed by others
Main
Bacterial meningitis remains a serious problem in contemporary medicine despite advances in intensive care and the use of adjunctive drugs to attenuate the host inflammatory response(1–4). In the past several years, the molecular mechanisms of the inflammatory cascade within the CNS have been well characterized and have been found to include release or generation of arachidonic acid metabolites, cytokines, nitric oxide, and radicals of molecular oxygen(5–12). Modulation of this complex inflammatory response serves as the basis for the use of antiinflammatory glucocorticoids to modulate disease severity(3, 4, 13). However, even with the use of glucocorticoids, there remains a substantial percentage of children with bacterial meningitis who suffer serious neurologic sequelae, suggesting that additional forms of modulation of the host response may be needed to further improve outcome(3, 4, 14). In addition to the proinflammatory and cytotoxic potential of these humoral mediators, these compounds may also have effects on the cerebral vascular bed and have direct effects on cellular metabolism. Compared with our understanding of the inflammatory events in the CNS in meningitis, less is known about the mechanisms underlying intracranial pathophysiologic abnormalities such as cerebral ischemia, cerebral anaerobic metabolism, and intracranial hypertension. The present study employs a noninvasive technique, NIRS, to determine changes in brain tissue Hb, Hbo2, dHb, and oxidized cerebral cytaa3 in rabbits with experimental meningitis.
METHODS
New Zealand White rabbits (2.0-2.5 kg) of both sexes were used. Rabbits were fed a standard diet and were housed in accordance with the guidelines of the Committee on Care and Use of Laboratory Animal Resources, the National Research Council. All studies were reviewed and approved by the Animal Research Committee of the University of California San Francisco. Rabbits were anesthetized with i.v. urethane (Fluka Chemie AG, Buchs, Switzerland) 2.0 g/kg and given supplemental doses (0.5 g/kg) as needed to maintain light general anesthesia. A peripheral i.v. line, right carotid artery line, and femoral artery line were placed as previously described(15–17).
Meningitis was induced by direct intracisternal injection of ≈1 × 106 colony-forming units of Streptococcus pneumoniae, type 3, obtained from a clinical isolate. CSF sampling was subsequently performed by direct cisternal puncture.
Arterial blood gas measurements were made in a portable blood gas laboratory (ABL2, Radiometer, Copenhagen). CSF lactate and glucose were measured in an autoanalyzer (YSI 2300 G/L, Yellow Springs Instrument Co., Yellow Springs, OH).
CBF was measured by the reference method(18) using labeled 15-μm latex microspheres, after dissection of the brain into left and right hemispheres, brainstem, and cerebellum. Calculation of CBF was determined by the formula: CBF = RBF ×Cx/Carterial where RBF is the withdrawal rate of the reference arterial blood sample,Cx is fluorescencer per 100 g of brain tissue, andCarterial is fluorescence in the reference arterial blood sample.
NIRS measurements were made with an LIA-4 research NIROSCOPE™ monitor(Vander Corp., Durham, NC). Operating principles of this instrument have been previously described in detail(19). Briefly, the instrument utilizes four continuous-wave laser diodes which emit in four wavelengths (781, 813.5, 860.5, and 917 nm) chosen to optimize resolution of oxidized cytaa3, Hbo2, and dHb. The transmission of NIRS signal through tissue is compared with baseline levels established by calibration at each wavelength, then converted by computer algorithms into a measured signal relative to the baseline signal. The signal is expressed in units ofv/d, where each unit is equivalent to a 10-fold change in transmitted signal. NIRS optrodes were positioned 3.0 cm apart, caudal to the orbits, and held in a polyvilylsiloxasone helmet to assure reproducible positioning for repeat measurements.
Data were analyzed by unpaired t test for comparison between groups and by ANOVA for repeated measurements within groups. Results were considered significant when P ≤ 0.05.
Experimental protocol. Rabbits were anesthetized, intravascular catheters inserted, and the animals were allowed to rest for 3-4 h before measurements were made. Baseline CBF and NIRS measurements were determined before infection. Rabbits were then randomly selected and infected by intracisternal injection of viable S. pneumoniae or saline vehicle. Repeat measurements for arterial blood gas, CSF parameters, CBF, and NIRS were performed at 18 and 22 h. Animals were killed at the conclusion of the study by injection of pentobarbital (150 mg/kg i.v.).
RESULTS
Six control and seven infected rabbits were studied. Before infection, baseline measurements of blood gases, CBF, and NIRS measurements were performed in three animals in each group and repeated at 18 and 22 h; in the remaining animals, measurements were performed at 18 and 22 h.
Arterial blood gases differed between control and infected rabbits at 18 and 22 h. Infected rabbits hyperventilated compared with controls with significant reduction in Pco2, increased Pao2 and oxygen saturation, and increased pH (Table 1). These findings are comparable to what has been reported previously in spontaneously breathing rabbits with experimental meningitis(17).
CSF examination demonstrated significant lactic acidosis(Table 1) and pleocytosis (data not shown) in infected animals compared with controls, which progressed between 18 and 22 h. CSF glucose was lower in infected animals; however, this finding did not reach statistical significance.
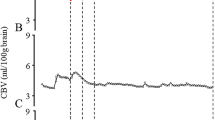
CBF was significantly reduced in infected animals compared with controls by 18 h (62.0 ± 13.0 versus 87.8 ± 10.9 mL/min/100 g,p < 0.05, t test) and was further reduced by 22 h (43.5± 31.8 versus 77.7 ± 16.9 mL/min/100 g, p< 0.05, t test) (Fig. 1).

CBF (mL/min/100 g) in control rabbits (open circles) and rabbits with meningitis (closed circles). Data are presented as mean ± SD. There is significant reduction in CBF at 18 and 22 h (p < 0.05, t test).
NIRS measurement revealed that infected rabbits had reduced brain tissue oxidized cytaa3 concentration compared with control rabbits. The reduction was progressive and significant at both 18 and 22 h(Fig. 2). Parameters of intracranial blood, dHb, and Hbo2 concentration showed a significant increase by 22 h in dHb in infected rabbits, which was not seen in controls (p = 0.04, ANOVA for repeated measures), and a trend toward decreased Hbo2 in infected rabbits compared with controls (p = 0.056, t test)(Figs. 3 and4). Total tissue Hb was not different between the two groups (Fig. 5).

Cytaa3 concentration (v/d units) in control rabbits (open circles) and rabbits with meningitis(closed circles). Data are presented as mean ± SE. Significant reduction of oxidized cytaa3 is present in rabbits with meningitis at 18 and 22 h (p < 0.05 at 18 h and < 0.005 at 22 h; t test).

dHb (v/d units) in control rabbits(open circles) and rabbits with meningitis (closed circles). Data are presented as mean ± SE. There is a progressive increase in dHb in rabbits with meningitis (p = 0.04, ANOVA for repeated measures) that is not seen in controls.

Hbo2 (v/d units) in control rabbits(open circles) and rabbits with meningitis (closed circles). Data are presented as mean ± SE. There is a trend toward reduced cerebral Hbo2 in infected rabbits compared with controls(p = 0.056, t test) at 18 h of infection.

THb (v/d units) in control rabbits(open circles) and rabbits with meningitis (closed circles). Data are presented as mean ± SE. No differences are present between control and infected rabbits.
DISCUSSION
The principal findings in this study were that brain tissue oxidized cytaa3 concentration was significantly decreased, and there was a relative reduction in Hbo2 accompanied by a significant increase in dHb in the brain in rabbits with meningitis. CBF was reduced in animals in the latter stages of experimental meningitis, as previously reported(16, 17, 20), and total brain Hb (THb) was not different between infected and control rabbits. Taken together, these results provide insight into changes in cerebral metabolism and cerebrovascular status of the brain in meningitis.
Cytaa3 is the final electron acceptor in the group of mitochondrial enzymes which constitute the respiratory chain. Electrons pass along the chain in a series of redox reactions, where with passage of electrons, the electron donor becomes oxidized, and the recipient becomes reduced. Oxidation of cytaa3 occurs with passage of electrons from cytaa3 to oxygen which completes mitochondrial oxidative phosphorylation with generation of ATP. Reduction in the tissue concentration of oxidized cytaa3, therefore, could represent either an inability of cytaa3 to react with oxygen, because of reduced oxygen availability as occurs in hypoxia or ischemia or an increase in the flow of electrons to cytaa3, as occurs with uncoupling of oxidative phosphorylation. Reduced cytaa3 has been demonstrated in experimental models of hypoxia(21–24) or hypoxia/ischemia(25), and in these settings it is presumed that the oxidized cytaa3 concentration is reduced because of reduced tissue oxygen delivery. Reduction in cerebral cytaa3 has also been described in one model of infection in which endotoxin was injected i.v. in the rat; however, the authors speculated that this result may have been due to cerebral ischemia secondary to systemic hypotension, rather than indicating a direct effect on the brain(26). The same group did, however, in a different study demonstrate reduction in intestinal oxidized cytaa3 in the absence of either ischemia or reduction in intestinal Hbo2 after endotoxin infusion in the rat. The presence of apparent energy failure, without substrate limitation, led the authors to speculate that it may have been due to endotoxin-induced uncoupling of oxidative phosphorylation(27).
In the present study, the mechanism of reduced cerebral cytaa3 could be due to cerebral ischemia in the infected group. CBF was reduced by approximately 40% at 22 h of infection, and it is possible that there was increased metabolic demand imposed by fever or another aspect of the infection which would potentiate the effect of ischemia. It is not as clear that the degree of ischemia present at 18 h of infection (roughly 25% compared with controls) would explain the significant reduction in cerebral oxidized cytaa3 present at that time. Lubarsky et al.(28) demonstrated in a rabbit model of normovolemic hemodilution that cerebral oxidized cytaa3 concentrations were unchanged when oxygen delivery was reduced to a level comparable to that at 18 h in our study. At that level of ischemia or reduced oxygen-carrying capacity, other compensatory mechanisms such as increased oxygen extraction could compensate for reduced substrate availability.
An alternative explanation for the reduction in oxidized cytaa3 is that meningitis may cause uncoupling of oxidative phosphorylation in mitochondrial enzymes involved in regeneration of oxidized cytaa3. It is known that IL-1β and TNF-α, released by the CNS in meningitis, are mediators of anaerobic metabolism in peripheral tissues in sepsis through induction of NO synthase and production of NO(29–31). Furthermore, we have previously shown that this effect is present in the CNS as well. In TNF-α-induced meningitis, cerebral oxygen uptake is reduced and cerebral anaerobic metabolism increased, and these effects are blocked by inhibition of NO production by the NO synthase inhibitor, L-NAME(6). NO and/or peroxynitrite, formed in the reaction of NO and oxygen radicals, are potent inhibitors of mitochondrial respiration through inhibition of the Krebs cycle enzyme, aconitase, and complexes I and II of the mitochondrial respiratory chain(32–37). Although not proven by the present study, it is possible that humoral factors may contribute to a shift to anaerobic metabolism which is additive to reduction in CBF. Further evidence that a humoral factor or factors is present in the CNS in meningitis is provided by earlier studies in our laboratory. We have previously reported that, although cerebral ischemia potentiates anaerobic metabolism in the CNS in meningitis, as evidenced by markedly increased CSF lactate with severe ischemia, CSF lactate is also significantly increased, although less so, under conditions of normal CBF in experimental meningitis(17).
The changes observed in intracranial blood (THb) and relative proportions of Hbo2 and dHb may also shed light on intracranial pathophysiology in meningitis. First, there was no significant difference in the brain tissue THb in rabbits with meningitis compared with controls. This is noteworthy, in that rabbits with meningitis had a reduction in CBF compared with controls and should, therefore, have had reduced THb. The reduction in CBF was accompanied by a decrease in tissue Hbo2 in infected animals, and an progressive and significant increase in dHb. The most likely explanation for comparable THb in the two groups is that, in the rabbits with meningitis, there was an increase in blood on the venous side of the cerebral capillary bed, which compensated for the reduction in blood on the arterial side. Although it is possible that the increase in dHb could be explained by increased oxygen extraction in animals with meningitis, we believe this is unlikely for two reasons. First, this would not account for the equivalence in THb between the two groups. In addition, in rabbits with TNF-α-induced meningitis, oxygen extraction fraction was reduced compared with controls(6).
The significance of an increase in intracranial venous blood could be of physiologic importance in that it could contribute to intracranial hypertension. We have previously found in experimental meningitis that increased ICP correlated with increased CBF(16) and that observation has subsequently been validated by Pfister et al.(11). These studies established that an increase in intracranial blood volume could increase ICP in this disease. In addition, in the model of TNF-α-induced meningitis, we showed that treatment with L-NAME inhibited increases in ICP compared with untreated animals at comparable levels of CBF, indicating that there is an NO-mediated component to increased ICP, and hypothesized that this was most likely to represent an increase in blood in the venous compartment(6). The current finding that there is an increase in intracranial dHb supports the hypothesis that cerebral venous engorgement is present in meningitis. Determination of the mechanism of this abnormality could lead to specific therapy to reduce the contribution of vascular engorgement to intracranial hypertension.
Abbreviations
- NIRS:
-
near-infrared spectroscopy
- THb:
-
total Hb
- dHb:
-
deoxygenated Hb
- Hbo2:
-
oxygenated Hb
- cytaa3:
-
cytochromeaa3
- CBF:
-
cerebral blood flow
- CSF:
-
cerebrospinal fluid
- ANOVA:
-
analysis of variance
- TNF:
-
tumor necrosis factor
- L-NAME:
-
NG-nitro-L-arginine methyl ester
- ICP:
-
intracranial pressure
- v/d :
-
variation in density
- Pao2:
-
partial pressure of arterial O2
- NO:
-
nitric oxide
References
Dodge PR, Hallowell D, Feigin RD, Holmes SJ, Kaplan SL. Jubelirer DP, Stechberg B. Hirsh SK 1984 Prospective evaluation of hearing impainment as a sequela of acute bacterial meningitis. N Engl J Med 311: 869–874.
Pomeroy SL, Holmes SJ, Dodge PR, Feigin RD 1990 Seizures and other neurologic sequelae of bacterial meningitis in children. N Engl J Med 323: 1651–1657.
Odio CM, Faingezicht I, Paris M, Nassar M, Baltodano A, Rodgers J, Saez-Llorens X, Olsen KD, McCracken GH Jr 1991 The beneficial effects of early dexamethasone administration in infants and children with bacterial meningitis. N Engl J Med 324: 1525–1531.
Label MH, Freij BJ, Syrogiannopoulos GA, Chrane DF, Hoyt MJ, Stewart SM, Kennard BD, Olson KD, McCracken GH Jr 1988 Dexamethasone therapy for bacterial meningitis. Results of two double-blind, placebo-controlled trials. N Engl J Med 319: 364–371.
Saez-Llorens X, Ramilo O, Mustafa MM, Mertsola J, McCracken GH Jr 1990 Molecular pathophysiology of bacterial meningitis: current concepts and therapeutic implications. J Pediatr 116: 671–684.
Tureen JH 1995 Effect of recombinant human tumor necrosis factor-α on cerebral oxygen uptake, cerebrospinal fluid lactate, and cerebral blood flow in the rabbit: role of nitric oxide. J Clin Invest 95: 1086–1091.
Tuomanen E, Liu H, Hengstler B, Zak O, Tomasz A 1985 The induction of meningeal inflammation by components of the pneumococcal cell wall. J Infect Dis 152: 859–868.
Tuomanen E, Tomasz A, Hengstler B, Zak O 1985 The relative role of bacterial cell wall and capsule in the induction of inflammation in penumococcal meninigitis. J Infect Dis 151: 535–540.
Tureen JH, Täuber MG, Sande MA 1991 Effect of indomethacin on the pathophysiology of experimental meningitis in rabbits. J Infect Dis 163: 647–649.
Quagliarello V, Scheld WM 1992 Bacterial meningitis: pathogenesis, pathophysiology, and progress. N Engl J Med 327: 864–872.
Pfister HW, Koedel U, Harbel RL, Dirangl U, Feiden W, Kuckdesche LG, Einhaupl KM 1990 Microvascular changes during the early phase of experimental bacterial meningitis. J Cereb Blood Flow Metab 10: 914–922.
Saukkonen K, Sadne S, Cioffe C, Wolpe S, Sherry B, Cerami A, Tuomanen E 1990 The role of cytokines in the generation of inflammation and tissue damage in experimental gram-positive meningitis. J Exp Med 171: 439–448.
Mustafa MM, Ramilo O, Oslen KD, Franklin PS, Hansen EJ, Beutler B, McCracken GH Jr 1989 Tumor necrosis factor α in mediating experimental Haemophilus influenzae type b meningitis. J Clin Invest 84: 1253–1259.
Wald ER, Kaplan SL, Manson EO Jr, Sabo D, Ross L, Arditi M, Wiedermann BL, Barson W, Kim KS, Yogev R, Hofkosh D, Meningitis Study Group 1995 Dexamethasone therapy for children with bacterial meningitis. Pediatrics 95: 21–28.
Dacey RG, Sande MA 1974 Effect of probenecid on cerebrospinal fluid concentrations of penicillin and cephalosporin derivatives. Antimicrob Agents Chemother 6: 437–441.
Tureen JH, Dworkin RJ, Kennedy SL, Sachdeva M, Sande MA 1990 Loss of cerebrovascular autoregulation in experimental meningitis in rabbits. J Clin Invest 85: 577–581.
Tureen JH, Täuber MG, Sande MA 1992 Effect of hydration status on cerebral blood flow and cerebrospinal fluid lactic acidosis in rabbits with experimental meningitis. J Clin Invest 89: 947–953.
Heymann MA, Payne BD, Hoffman JIE, Rudolph AM 1977 Blood flow measurements with radionuclide-labeled particles. Prog Cardiovasc Dis 20: 55–70.
Jöbsis F 1977 Noninvasive monitorin of cerebral and myocardial oxygen sufficiency and circulation parameters. Science 198: 1264–1267.
Täber MG, Burroughs M, Niemöller UM, Kuster H, Broschberg U, Tuomanen E 1991 Differences of pathophysiology in experimental meningitis caused by three strains of Streptococcus pneumoniae. J Infect Dis 163: 806–811.
Thornily MS, Livera N, Wickramasinghe YABD, Rolfe P 1990 A study in vivo into the kinetics of the dissociation of oxygen from oxyhaemoglobin compared with changes in the redox state of cytochrome oxidase in rat brain utilizing near-i.r. spectroscopy. Biochem Soc Trans 18: 1019–1020.
Tsuji M, Naurse H, Volpe J, Holtzman D 1995 Reduction of cytochrome aa3 measured by near-infrared spectroscopy predicts cerebral energy loss in hypoxic piglets. Pediatr Res 37: 253–259.
Hampson NB, Camporesi EM, Stolp BW, Shook JE, Griebel JA, Piantodosi CA 1990 Cerebral oxygen availability by NIR spectroscopy during transient hypoxia in humans. J Appl Physiol 69: 907–913.
Lubrasky DA, Griebel JA, Carnporesi EM, Piantodosi CA 1992 Comparison of systemic oxygen delivery and uptake with NIR spectroscopy of brain during normovolemic hemodilution in the rabbit. Resuscitation 23: 45–57.
Piantodosi CA, Helmstreet TM, Jöbsis-VanderVliet FF 1986 Near-infared spectrophotometric monitoring of oxygen distribution to intact brain and skeletal muscle tissues. Crit Care Med 14: 698–706.
Schaefer CF, Biber B 1993 Effects of endotoxemia on the redox state of brain cytoschrome a, a3 in rats. Circ Shock 40: 1–8.
Schaefer CF, Lerner MR, Biber B 1991 Dose-related reduction of intestinal cytochrome aa3 induced by endotoxin in rats. Circ Shock 33: 17–25.
Lubarsky DA, Griebel JA, Carnporesi EM, Piantodosi CA 1992 Comparison of systemic oxygen delivery and uptake with NIR spectroscopy of brain during normovolemic hemodilution in the rabbit. Resuscitation 23: 45–57.
Natason C, Eichenholz P, Danner R 1989 Endotoxin and turnor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J Exp Med 169: 823–832.
Nussler AK, Di Silvio M, Billiar TR, Hoffman RA, Geller DA 1992 Stimulation of the nitric oxide synthase pathway in human hepatocytes by cytokines and endotoxin. J Exp Med 176: 261–264.
Kilbourn RG, Gross SS, Jurban A, Adams J, Griffith OW, Levi R, Lodato RF 1990 NG-Methyl-L-arginine inhibits tumor necrosis factor-induced hypotension: implications for the involvement of nitric oxide. Proc Natl Acad Sci USA 87: 3629–3632.
Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH 1994 Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FFBS Lett 345: 50–54.
Tucker SD, Auzenne EJ, Sivaramakrishnan MR 1993 Inhibition of tumor cell mitochondrial respiration by macrophage cytoxic mediators distinct from interferon-γ. J Leukocyte Biol 53: 138–143.
Geng Y, Hansson GK, Holme E 1992 Interferon-γ and tumor necrosis factor synergize to induce nitric oxide production and inhibit mitochondrial respiration in vascular smooth muscle cells. Circ Res 71: 1268–1276.
Stadler J, Billiar TR, Curran RD, Stuehr DJ, Ochoa JB, Simmons RL 1991 Effect of exogenous and endogenous nitric oxide on mitochondrial respiration of rat hepatocytes. Am J Physiol 260:C910–C916.
Welsh N, Sandler S 1992 Interleukin-1β induces nitric oxide production and inhibits the activity of aconitase without decreasing glucose oxidation rates in isolated mouse pancreatic islets. Biochem Biophys Res Commun 182: 333–340.
Radi R, Rodriguez M, Castro L, Telleri R 1994 Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 308: 89–95.
Acknowledgements
The authors thank the University of California San Francisco Academic Senate Bachrach Fund for the purchase of the NIRS system, and Frans JöbsisVanderVliet, Ph.D., for his critical review and suggestions.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Tureen, J., Liu, Q. & Chow, L. Near-Infrared Spectroscopy in Experimental Pneumococcal Meningitis in the Rabbit: Cerebral Hemodynamics and Metabolism. Pediatr Res 40, 759–763 (1996). https://doi.org/10.1203/00006450-199611000-00016
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199611000-00016
