
Abstract
Three patients with propionic acidemia were studied. The first patient was diagnosed at the age of 9 mo, 3 mo after he developed hypotomia and choreoathetoid movements after an upper respiratory tract infection. The second patient was diagnosed at the age of 1.5 mo when she became comatose after nasogastric tube feeding because of failure to thrive. The third patient was diagnosed at the age of 5 d when she presented with feeding difficulties, hypotonia, and respiratory insufficiency. Magnetic resonance imaging (MRI) of the brain in all patients revealed delayed myelination and some cerebral atrophy. In the patient with choreoathetosis, MRI showed bilateral abnormalities in the signal intensity of the putamen and caudate nuclei. MRI of the other two patients showed normal basal ganglia. Proton magnetic resonance spectroscopy (1H MRS) from a voxel located in the basal ganglia revealed a decrease in N-acetylaspartate and myo- inositol peaks and an elevation of glutamine/glutamate. The presence of spectroscopic abnormalities in a stable metabolic condition, in particular the rise in glutamine/glutamate, indicates that the metabolic balance on cerebral parenchymal level is less optimal than estimated from biochemical analysis of urine, plasma, or cerebrospinal fluid.
Similar content being viewed by others
Main
Propionic acidemia is a genetic disorder of amino acid metabolism caused by a deficiency of propionyl-CoA carboxylase(1–3). The first clinical presentation may be manifest at any age from the neonatal period on. The spectrum of possible clinical symptoms is wide. Many patients exhibit poor feeding, vomiting, and lethargy leading to coma if onset of treatment is delayed. Metabolic derangement may be precipitated by infection or excessive protein intake(4). Ketoacidosis and hyperammonemia are prominent laboratory findings, in particular during episodes of metabolic decompensation. The reported neurologic symptomatology is diverse. Frequently observed neurologic signs are axial hypotonia, extrapyramidal manifestations, and seizures (see Table 1). Surtees et al.(5) recently reported the neurologic outcome of 20 patients with propionic acidemia. Extrapyramidal movements were a prominent finding in both the early-onset group and the late-onset group (Table 1). In the late-onset group, bilateral basal ganglia hypodensities were observed on cranial computer tomography when performed shortly after the onset of the movement abnormalities. These lucencies resolved completely in most patients over a 1-3-mo period after the onset of dietary treatment(5). The pathophysiology of these basal ganglia abnormalities is unknown.
We report proton (1H) MRS studies of the basal ganglia in three patients with propionic acidemia (one with an early onset, two with the late-onset form). The aim of the study was to investigate whether 1H MRS may contribute to the insight of the underlying mechanisms accounting for the basal ganglia damage in propionic acidemia and whether 1H MRS may contribute in the monitoring of treatment.
METHODS
Three children with propionic acidemia were studied. The diagnosis was made on the basis of clinical presentation, urinary metabolite excretion pattern, and in vitro enzyme assays. Propionyl-CoA carboxylase activity in cultured skin fibroblasts ranged from 0.2 to 1 nmol/h/mg protein (normal 15-75). The clinical symptoms are summarized in Table 1. Patient 1 presented at the age of 6 mo with hypotonia and choreoathetoid movements and was diagnosed at 9 mo of age. Patient 2 came to attention at the age of 1.5 mo when she became comatose after nasogastric tube feeding because of failure to thrive. Patient 3 was diagnosed at the age of 5 d when she presented with feeding difficulties, hypotonia, and respiratory insufficiency. Patients 2 and 3 required hemodialysis or peritoneal dialysis. Once the diagnosis was made, all of the patients were treated with a low protein diet and were given a trial of biotin, which was discontinued when they were shown to be unresponsive. Patient 1 was followed up to his present age of 36 mo and is still severely retarded and choreoathetotic. Patient 2 presented a normal development at the age of 3 mo. Patient 3 had initially repeated episodes of metabolic decompensation, but has remained stable now for 1 y. At 32 mo of age she was still retarded, but she had improved considerably during the last year. At no time choreoathetosis was noted. MRI and 1H MRS of the brain were performed in all three patients during stable clinical and biochemical conditions.
Organic acids in plasma, urine, and CSF were quantitated by gas chromatography(6). A standard amino acid analyzer was used to determine the amino acids in plasma, urine, and CSF. CSF concentrations of free and total GABA were determined by using a stable isotope dilution gas chromatography/mass spectrometry assay(7).
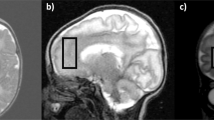
MRI and MRS were performed on a 1.5 Tesla MR instrument. Sagittal T1-weighted images and transverse T1- and T2-weighted images were made. The standard imaging head-coil was used for 1H MRS. A 2 × 2 × 2-cm voxel was chosen in the area of the caudate nucleus, putamen, globus pallidus, and thalamus on the left side, containing a mixture of white and, in particular, central gray matter. The STEAM sequence was used with a repetition time of 2500 ms, an echo time of 20 ms, and 128 acquisitions[see Frahm et al.(8) for all technical details]. The averaged measurements were zero-filled to 2048 data points, exponentially filtered to give 1 Hz line-broadening before fast-Fourier transformation. The spectra were quantified by peak area measurements using system software (Siemens SP63). The following peaks were quantified: NAA (2.02 ppm), Glx (2.1-2.5 ppm), “creatine” representing creatine and phosphocreatine (Cr, 3.02 ppm), “choline” representing choline-containing compounds (Cho, 3.20 ppm). and mI (3.56 ppm). To determine a baseline, four fixed areas were used: left foot of the NAA peak (2.02 ppm), area between the Cr peak (3.02 ppm) and the NAA peak at 2.70 ppm, left foot of the mI peak (3.56 ppm), and the area at the right of the Cr peak at 3.93 ppm. The peaks of spectra were normalized to the Cr peak (3.02 ppm). Creatine age- and area-dependent values were used as known from the literature(9, 10). An exception was made for the region between 2.1 and 2.5 ppm, containing multiple overlapping peaks representing glutamine, glutamate, and some GABA (together Glx). The ratio of the integral of this area and the creatine peak was used as a rough indication of the concentration of the contributing metabolites. The control group for the MRS investigations consisted of eight healthy children varying in age from 3 to 48 mo.
RESULTS
Results of biochemical studies are summarized in Table 2. In the period of the 1H MRS all patients had normal plasma ammonia values. The urinary 3-OH-propionic acid concentration was elevated in all patients. The free and total GABA in CSF was normal, whereas CSF glycine and alanine concentrations were increased in all patients.
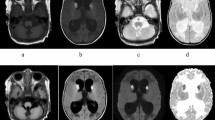
Initial MRI of all patients showed a mildly enlarged ventricular system and subarachnoid spaces. The MRI of patient 1 at 10 mo of age (Fig. 1) showed an abnormally high signal intensity on T2-weighted images, abnormally low on T1-weighted images in the caput and corpus of the caudate nucleus and in the putamen. Globus pallidus and thalamus were normal. Myelination of the cerebral hemispheres was delayed and irregular with patchy areas of high signal intensity on T2-weighted images and low signal intensity on T1-weighted images. At the age of 24 and 36 mo the repeat MRIs showed progress in myelination and diminishing cerebral atrophy, but the basal ganglia abnormalities remained unchanged.

MRI of the brain of patient 1 at the age of 10 mo. The T2-weighted MR images show enlarged subarachnoid spaces and ventricular system, lesions in the putamen and caudate nucleus, and delayed myelination.
MRI of patient 2, made at the age of 2 mo, showed, apart from some cerebral atrophy, no other abnormalities. The images showed the beginning of myelination and normal basal ganglia.
At 20 mo of age the MRI of patient 3 showed mildly delayed myelination, which was not yet completed in the temporal lobes and in the directly subcortical areas. Some mild and patchy increases in signal intensity were seen in the frontal and occipital periventricular white matter on T2-weighted images. The basal ganglia were normal. Repeat MRI at the age of 32 mo showed considerable improvement of myelination and diminished cerebral atrophy.
The spectroscopic data from the area of the basal ganglia of the patients were compared with those of the eight healthy controls. The data are summarized in Table 3 (see also Fig. 2). In all patients the level of NAA was decreased. The Cho level in patients 1 and 3 did not differ from the level in controls, but was elevated in patient 2. In patients 1, 2, and to a lesser extent in patient 3, elevated signals were seen in the range of 2.1-2.5 ppm, indicating a rise in Glx. In all patients mI was decreased.

Short echo time STEAM spectrum obtained from the area of the basal ganglia on the left side in patient 2 (upper spectrum), compared with a normal spectrum of a control child of the same age(lower spectrum). Note the decrease in NAA (2.02 ppm), increase in Glx (2.1-2.4 ppm), rise in Cho (3.2 ppm), and decrease in mI (3.56 ppm) relative to Cr (3.02 ppm) in patient 2 compared with the control.
DISCUSSION
Propionic acidemia is caused by a defect in the carboxylation of propionyl-CoA to methylmalonyl-CoA. Biochemically this disorder is characterized by moderate to severe ketoacidosis, hyperglycinemia, and hyperammonemia. The acidosis is the result of accumulation of various normal and abnormal organic acids, in particular propionate, 3-hydroxybutyrate, methylcitrate, and 3-hydroxypropionate. Propionate is derived from hydrolysis of propionyl-CoA and is detectable in all body fluids. The hyperammonemia during acute episodes is thought to be caused by the inhibitory effects of propionyl-CoA on N-acetylglutamate synthetase, the first enzyme involved in ureagenesis(11).
Although the metabolic routes of the production of abnormal acids in propionic acidemia are known(12) and the effects of these organic acids on different organ systems have been studied, the explanation for the heterogeneous clinical presentation is unknown(13). The differentiation between the early-onset and late-onset group can neither be made on the basis of different genetic mutations(14), nor on the basis of different levels of residual activity of propionyl-CoA carboxylase(13). In general, episodes of clinical deterioration are accompanied by metabolic decompensation, but clinical deterioration and necrosis of the basal ganglia have been described despite documented excellent metabolic control at the time of onset of the cerebral dysfunction(15).
Our three patients had different clinical presentations. Patient 1 presented with choreoathetosis at the age of 6 mo. Despite adequate treatment the choreoathetosis persisted, probably because of irreversible damage of the basal nuclei. The delay in treatment may be important in this respect. MRI of the brain at the age of 10, 24, and 36 mo showed symmetrical lesions in the putamen and caudate nucleus, in addition to a delay in myelination and some cerebral atrophy. Patients 2 and 3 presented clinical symptoms at 1.5 mo and 5 d, respectively. They had no choreoathetosis, nor abnormalities of the basal nuclei.
Bilateral lesions of the basal nuclei have been often noted in propionic acidemia, in particular in the late-onset variant (Table 1). The pathophysiologic mechanisms underlying these lesions, however, are still unknown.
The MRS findings from the basal ganglia in our patients were essentially identical showing decreased NAA, increased Glx, and decreased mI peaks in comparison with the values found in healthy controls. These changes were reproducible in follow-up. The decreased level of NAA is commonly ascribed to neuronal dysfunction, damage, or loss(16). It is possible that the decrease in NAA can be partially ascribed to delayed neuronal maturation.
At the lower magnetic field strengths used in clinical studies, the separation of glutamine from glutamate is not possible. The elevation of Glx may represent an increase in glutamine(17–20). Elevated glutamine peaks in MRS of the brain have been described most often in hyperammonemia(17–20). Hyperammonemia leads to enhanced production of glutamine and glutamate from 2-oxoglutarate. Glutamate is a major excitatory neurotransmitter, whereas GABA, formed by decarboxylation from glutamate, is the most important inhibitory neurotransmitter. In the presynaptic neuron, glutamine is converted to glutamate and ammonia by glutaminase. Glutamate can either be converted to GABA by glutamic acid decarboxylase or be released in the synaptic cleft. After release by the presynaptic neuron, glutamate is taken up by the astrocyte where it is converted into glutamine by glutamine synthetase and then transported back to the presynaptic neuron. This cycle operates mainly between the presynaptic neuron, the neuronal cleft, and the astrocyte. Hyperammonemia has a great impact on this cycle by stimulating glutamine synthesis via glutamine synthetase, by possible inhibition of glutaminase, and by inhibition of glutamate re-uptake by the astrocyte. High plasma ammonia levels would lead to an accumulation of glutamine in the brain, mainly in astrocytes. An imbalance between excitatory and inhibitory neurotransmitters may contribute to the cerebral dysfunction. Accumulation of intracellular glutamine is reflected in elevated Glx in 1H MRS(17–20). However, at the time of1 H MRS in our three patients, the plasma ammonia concentration and the plasma and CSF concentrations of glutamine and glutamate were normal or just above normal (see Table 2). This indicates that blood and CSF concentrations may differ from tissue concentrations. Differences between blood, CSF, and brain parenchyma metabolite concentrations have been reported before in metabolic disorders, in particular with respect to lactate in mitochondrial disorders(21, 22). MRS may reveal considerable elevation of lactate in brain tissue, whereas lactate in CSF and blood is normal or only slightly elevated(21). On the other hand, MRS may not show any evidence of elevated lactate, whereas CSF lactate is high(22). Assessment of brain tissue metabolite concentrations by MRS is an additional method for monitoring metabolic stability at tissue level to the biochemical analysis of urine, plasma, and CSF.
Glutamate is a major excitatory neurotransmitter in the CNS and is well known for its excitotoxic effects in many forms of brain damage, particularly in hypoxia-ischemia. Elevated Glx reflecting elevated glutamate has been found in cases of acute cerebral hypoxia-ischemia(23–25). The elevated Glx that we found in our patients could be a sign of active excitotoxic damage of the basal nuclei. Considering the clinical and metabolic stability of the patients at the time of MRS, this hypothesis is less attractive.
Another possible explanation for basal nuclei dysfunction in propionic acidemia could be alterations in mitochondrial energy metabolism causing accumulation of toxic metabolites(15). The presence of elevated cerebral lactate levels is an argument in favor of this hypothesis. However, although we found slightly increased lactate values in CSF, we found no spectroscopic evidence of increased lactate within the brain in our patients.
It has been suggested that one or more of the metabolites accumulating in propionic acidemia is neurotoxic(15). Our data show that 3-OH-propionate is the only metabolite of the propionate derivatives detectable in large quantity in CSF. Unfortunately this metabolite could not be detected by MRS methods.
Reduced cerebral mI as found in our patients has also been observed in patients with hyperammonemia and elevated Glx levels. Glycine co-resonates with mI at 3.56 ppm(26). CSF glycine was elevated in all patients. It cannot be excluded that mI was actually lower than now estimated, because the decrease in peak area was partly compensated by the increase in glycine. However, it is not probable that there was a major contribution of glycine in our patients. In all patients additional spectra were acquired with a longer echo time (135 ms). These did not show the presence of a resonance at 3.56 ppm, which would have been expected in the case of elevation of glycine. The significance of the mI reduction is not understood. It is generally believed that mI plays a role as a precursor of the inositol polyphosphate family of intracellular messengers. It may play an important role in the detoxification reactions of the brain(17, 18), and it has been suggested that low mI may be the result of enhanced use of detoxification processes. Another function of mI may be that of osmoregulator(17, 18). It is possible that the decrease in mI is compensatory for the increase in Glx to maintain osmotic balance in the brain.
The observation of spectroscopic abnormalities in metabolically stable patients is important. Serious, even fatal, neurologic deterioration with basal ganglia necrosis may occur in patients who are in a stable metabolic condition as estimated by conventional biochemical analyses(15). The current study shows that biochemical changes at tissue level as seen by MRI/MRS differ from the results obtained by biochemical analysis of body fluids. MRS of the brain in vivo would provide a valuable, noninvasive tool in monitoring patients with propionic acidemia and other inborn errors of organic acid metabolism. In this respect MRI and MRS would have complementary values, MRI reflecting both temporary and permanent cerebral structural damage, and MRS reflecting actual intracellular biochemical changes at tissue level.
Abbreviations
- Cho:
-
choline-containing compounds
- Cr:
-
creatine and phosphocreatine
- CSF:
-
cerebrospinal fluid
- Glx:
-
glutamine and glutamate
- mI:
-
myo-inositol
- MRI:
-
magnetic resonance imaging
- MRS:
-
magnetic resonance spectroscopy
- NAA:
-
N-acetylaspartate
- ppm:
-
parts per million
- GABA:
-
γ-aminobutyric acid
- STEAM:
-
stimulated-echo acquisition mode
References
Childs B, Nyhan WL, Borden M, Bard L, Cooke RE 1961 Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. Pediatrics 27: 522–538
Hommes FA, Kuipers JRG, Elema JD, Jansen JF, Jonxis JHP 1968 Propionic acidemia, a new inborn error of metabolism. Pediatr Res 2: 519–524
Fenton WA, Rosenberg LE 1995 Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease, 7th Ed. McGraw-Hill, New York, pp 1431–1434
Wolf B, Hsia EY, Sweetman L, Gravel R, Harris DJ, Nyhan WL 1981 Propionic acidemia: a clinical uptake. J Pediatr 99: 835–846
Surtees RAH, Matthews EE, Leonard JV 1992 Neurologic outcome of propionic acidemia. Pediatr Neurol 8: 333–337
Duran M, Bruinvis L, Ketting D, de Klerk JBC, Wadman SK 1988 Plasma cis-4-decenoic acid, a characteristic metabolite in medium-chain acyl-CoA dehydrogenase deficiency. Clin Chem 34: 548–551
Kok RM, Howells DW, Heuvel CCM, van den Guerand WS, Thompson GN, Jakobs C 1993 Stable isotope dilution analysis of GABA in CSF using simple solvent extraction and electron-capture negative-ion mass fragmentography. J Inherit Metab Dis 16: 508–512
Frahm J, Michaelis T, Merboldt KD, Bruhn H, Gyngell ML, Hänicke W 1990 Improvements in localized 1H NMR spectroscopy of human brain: water suppression, short echo times, and 1 mL resolution. J Magn Reson 90: 464–473
Toft PB, Leth H, Lou HC, Pryds O, Henriksen O 1994 Metabolite concentrations in the developing brain estimated with proton MR spectroscopy. J Magn Reson Imaging 4: 674–680
Kreis R, Ernst T, Ross BD 1993 Development of the human brain: in vivo quantification of metabolite and water content with proton magnetic resonance spectroscopy. Magn Reson Med 30: 424–437
Wolf B, Hsia YE, Tanaka K, Rosenberg LE 1978 Correlation between serum propionate and blood ammonia concentrations in propionic acidemia. J Pediatr 93: 471–473
Van Rooyen JPG, Mienie LJ, Erasmus E, de Wet WJ, Duran M, Wadman SK 1994 Urinary excretion of homocitric acid and methylhomocitric acid in propionic acidaemia: minor metabolic products of the citrate synthase aldol condensation reaction. Clin Chim Acta 230: 91–99
Sethi KD, Ray R, Roesel RA, Carter AL, Gallagher BB, Loring DW, Hommes FA 1989 Adult-onset chorea and dementia with propionic acidemia. Neurology 39: 1343–1345
Gravel RA, Lam KF, Scully KJ, Hsia YE 1977 Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet 29: 378–388
Haas RH, Marsden DL, Capistrano-Estrada S, Hamilton R, Grafe MR, Wong W, Nyhan WL 1995 Acute basal ganglia infarction in propionic acidemia. J Child Neurol 10: 18–22
Van der Knaap MS, van der Grond J, Luyten PR, den Hollander JA, Nauta JJP, Valk J 1992 1H and 31P magnetic resonance in spectroscopy of the brain in degenerative cerebral disorders. Ann Neurol 31: 202–211
Kreis R, Farrow N, Ross BD 1991 Localized 1H NMR spectroscopy in patients with chronic hepatic encephalopathy: analysis of changes in cerebral glutamine, choline and inositols. NMR Biomed 4: 109–116
Ross BD 1991 Biochemical considerations in 1H spectroscopy: glutamate and glutamine; myo-inositol and related metabolites. NMR Biomed 4: 59–63
Kreis R, Ross BD, Farrow NA, Ackerman Z 1992 Metabolic disorders of the brain in chronic hepatic encephalopathy detected with H-1 MR spectroscopy. Radiology 182: 19–27
Connelly A, Cross JH, Gadian DG, Hunter JV, Kirkham FJ, Leonard JV 1993 Magnetic resonance spectroscopy shows increased brain glutamine in ornithine carbamoyl transferase deficiency. Pediatr Res 33: 77–81
Krägeloh-Mann I, Grodd W, Schöning M, Marquard K, Nägele T, Ruitenbeek W 1993 Proton spectroscopy in five patients with Leigh's disease and mitochondrial enzyme deficiency. Dev Med Child Neurol 35: 769–770
Kruse B, Hanefeld F, Holzbach U, Wilichowski E, Christen HJ, Merboldt KD, Hänicke W, Frahm J 1994 Proton spectroscopy in patients with Leigh's disease and mitochondrial enzyme deficiency. Dev Med Child Neurol 36: 839–840
Choi DW 1988 Glutamate neurotoxicity and diseases of the nervous system. Neuron 1: 623–634
Higuchi T, Fernandez EJ, Shimizu H, Weinstein PR, Maudsley AA, Weiner MW 1991 Effects of focal brain ischemia on lactate,N- acetyl aspartate and glutamate. In: Book of Abstracts of Society of Magnetic Resonance in Medicine, Berkeley, p 145
Higuchi T, Fernandez EJ, Rooney WD, Gaspary HL, Graham SH, Weinstein PR, Maudsley AA, Weiner MW 1992 Regional specific changes of N- acetyl aspartate, lactate, alanine and glutamate in post globally ischemic brain. In: Book of Abstracts of Society of Magnetic Resonance in Medicine, Berkeley, p 547
Heindel W, Kugel H, Roth B 1993 Noninvasive detection of increased glycine content by proton MR spectroscopy in the brains of two infants with nonketotic hyperglycinemia. Am J Neuroradiol 14: 629–635
Barnes DM, Enzmann DR 1981 The evoluation of white matter disease as seen on computed tomography. Radiology 138: 379–383
Gebarski SS, Gabrielsen TO, Knake JE, Latack JT 1982 Cerebral CT findings in methylmalonic and propionic acidemias. Am J Neuroradiol 4: 955–957
Kurczynski TW, Hoppel CL, Goldblatt PJ, Gunning WT 1989 Metabolic studies of carnitine in a child with propionic acidemia. Pediatr Res 26: 63–66
Hoffmann GF, Meier-Augenstein W, Stöckler S, Surtees R, Rating D, Nyhan WL 1993 Physiology and pathophysiology of organic acids in cerebrospinal fluid. J Inherit Metab Dis 16: 648–669
Acknowledgements
The authors thank Dr. O. P. van Diggelen for the determination of the propionyl-CoA carboxylase activity in cultured skin fibroblast, Dr. C. Jakobs for the determination of free and total GABA in CSF, and Drs. J. B. C. de Klerk, Prof. Dr. U. Wendel, and Drs. A. M. Hemmes for referring the patients.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Bergman, A., Van Der Knaap, M., Smeitink, J. et al. Magnetic Resonance Imaging and Spectroscopy of the Brain in Propionic Acidemia: Clinical and Biochemical Considerations. Pediatr Res 40, 404–409 (1996). https://doi.org/10.1203/00006450-199609000-00007
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199609000-00007
This article is cited by
-
Pericytes in Neurometabolic Diseases
Current Tissue Microenvironment Reports (2020)
-
In Vivo NMR Studies of the Brain with Hereditary or Acquired Metabolic Disorders
Neurochemical Research (2015)
-
Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia
Orphanet Journal of Rare Diseases (2014)
-
Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients
Orphanet Journal of Rare Diseases (2013)
-
Amino acid metabolism in patients with propionic acidaemia
Journal of Inherited Metabolic Disease (2012)
