
Abstract
The expression of the CC chemokine receptor-7 (CCR7) by cancers, including melanoma, augments lymph node (LN) metastasis, but little is known about its role in lymphangiogenesis and anti-tumor immunity. We injected control B16 murine melanoma cells (pLNCX2-B16) and CCR7-overexpressing B16 cells (CCR7-B16) in murine footpads and compared resulting tumors at the protein and mRNA level using immunostaining, Affymetrix gene microarray and quantitative reverse-transcriptase PCR. Although control and CCR7-B16 primary tumors were of similar size, LN metastasis was dramatically enhanced in CCR7-B16 tumors. Microarray analysis of leukocyte-depleted pLNCX2-B16 and CCR7-B16 tumor cell suspensions showed that three major groups of genes linked to interferon (IFN)-γ signaling pathways (for example, STAT1, CXCR 9-11, CCL5 and CXCL10, major histocompatibility complex (MHC) I and MHC II) were downregulated in the CCR7-B16 tumor microenvironment, suggesting activation through CCR7 can downregulate pathways critical for host anti-tumor immunity. In addition, mRNA expression of the lymphatic marker podoplanin was upregulated in CCR7-B16 tumors by 3.35-fold versus control tumors. Anti-podoplanin monoclonal antibody staining revealed a three-fold increase in intratumoral CCL21-expressing lymphatic vessels, as well as a two-fold increase in the number of invading tumor cells per lymphatic vessel in CCR7-B16 versus control tumors. Enhanced anti-vascular endothelial growth factor C (VEGF-C) staining was present in CCR7-B16 versus control tumors, suggesting that VEGF-C may have a role in the CCR7-mediated lymphangiogenesis. In summary, CCR7-B16 tumors show a striking decrease in IFN-γ-mediated inflammatory gene expression in contrast to increased expression of VEGF-C, CCL21 and podoplanin by lymphatic vessels. Enhanced lymphangiogenesis may contribute to the dramatic increase in LN metastasis that is observed in the CCR7-expressing tumors.
Similar content being viewed by others


Introduction
The CC chemokine receptor-7 (CCR7) is expressed by many human cancers, including melanoma.1 In gastric cancer, CCR7 expression has been highly correlated with lymph node (LN) metastasis and invasion, as well as poor survival.2 Overexpression of CCR7 in B16 murine melanoma cells (pLNCX2-B16) increased the rate of metastasis to regional LNs by over 100-fold in experimental models of nodal metastasis.3 Recent data from our laboratory indicated overexpression of CCR7 in the B16 melanoma can also directly affect tumorigenesis, possibly by altering the host anti-tumor inflammatory response. Specifically, we showed injection of limiting numbers of CCR7-overexpressing B16 cells (CCR7-B16) into mouse ear skin yielded obvious tumors, whereas ear skin injected with control cells lacking CCR7 failed to develop tumors. When larger tumor inocula were used, however, the expression of CCR7 had no impact on tumor formation, but did result in markedly diminished trafficking of T cells and other inflammatory cells to the tumor microenvironment.4 Herein, to specifically determine alterations in the immune environment of CCR7-expressing and non-expressing tumors, we compared the mRNA expression of CCR7-B16 tumors and pLNCX2-B16 control tumors of approximately equal size. Before gene-expression profiling, we depleted CD45-expressing leukocytes from whole-tumor homogenates to remove profile bias resulting from the much larger numbers of leukocytes found in control tumors.4
CCR7-expressing tumor cells display enhanced metastasis to regional LN, presumably because CCR7 mediates chemoattraction between tumor cells and lymphatic endothelial cells that express CCR7 ligands, such as CCL21.5, 6 However, the effect of CCR7 expression on the tumor vasculature and microenvironment in facilitating nodal metastasis is unclear. From our profiling studies above, one gene that was overexpressed in CCR7-B16 tumors relative to the control tumors was podoplanin, a known specific marker for lymphatic endothelial cells.7 Using immunohistochemical staining approaches, we now report that podoplanin+ vessels are increased in CCR7-B16 tumors and that vascular endothelial growth factor C (VEGF-C), a lymphatic vessel growth factor,8 is also expressed at higher levels within the CCR7-B16 tumors.
Collectively, our data suggest newly formed CCR7-B16 tumors showed downregulation of interferon (IFN)-γ-associated genes, as well as major histocompatibility complex (MHC) class I and II molecules, suggesting early escape from anti-tumoral immunosurveillance. Strikingly, CCR7-B16 tumors exhibited increased recruitment of CCR7-B16 cells to podoplanin-expressing lymphatic vasculature. Together, these results suggest CCR7 modulates the tumor microenvironment by promoting early escape from immunosurveillance and increasing metastatic efficiency to LN via neo-lymphangiogenesis, with associated increases in chemotaxis and lymphatic invasion of tumor cells.
Results
Lack of early host anti-tumor immune responses in mouse CCR7-B16 footpad tumors
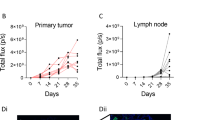
In our previous work, CCR7-B16 cells possessed a growth advantage over pLNCX2-B16 cells when small tumor cell inocula were used. Larger inocula developed into equivalent-sized footpad tumors. Interestingly, CCR7-B16 tumors are characterized by a marked reduction in the percentage of tumor-infiltrating leukocytes (especially CD8+ T cells) when compared with equivalently sized pLNCX2-B16 tumors.4
To investigate the host anti-tumor immune response generated in draining LN (DLN), we harvested popliteal DLN and non-draining contralateral popliteal LN, and measured the number of IFN-γ-producing cells by enzyme-linked immunosorbent spot (ELISPOT) assay. A robust host immune response was observed in DLN from pLNCX2-B16-injected mice at day 5, whereas only a minimal level of IFN-γ production was found in DLN from CCR7-B16-inoculated mice (Figure 1), suggesting a potential role for tumoral CCR7 expression in the suppression of early host anti-tumor immune response.

Downregulation of IFN-γ-producing cells in DLN of CCR7-B16 tumors. The numbers of cells producing IFN-γ in response to tumor cell stimulation in popliteal DLN were counted by ELISPOT assay on day 5, 11 and 21 after footpad inoculation; non-draining contralateral popliteal LNs (NDLNs) were used as control.
Identification of differentially expressed genes in CCR7-B16 tumors compared with pLNCX2-B16 tumors by Affymetrix microarray expression profiling
To understand the mechanism of immune suppression in CCR7-B16 tumors, we characterized the gene expression profile within the tumor microenvironment using Affymetrix expression microarrays. As the greater number of CD45+ leukocytes present in control tumors (versus CCR7-B16 tumors4) were a potential source of bias in gene-expression profiling, we depleted CD45+ cells using magnetic beads from single cell suspensions of both pLNCX2-B16 and CCR7-B16 tumors, resulting in >99% efficiency in removing CD45+ leukocytes. Three independent biological samples were prepared and analyzed with Affymetrix Mouse Genome 430 2.0 Arrays. Genomatix ChipInspector software was then used to identify genes showing significant and greater than 1.5-fold difference in mRNA expression between the CCR7-B16 and control tumors. Pathway analysis was carried out with the ChipInspector and BiblioSphere software to sort genes into groups according to known signaling or biological pathways.
Three groups of genes were significantly downregulated in CCR7-B16 tumors compared with pLNCX2-B16 tumors (Table 1). The first group included genes involved in the IFN-γ signaling pathways, as well as downstream IFN responsive genes, including transcription factors (STAT1, STAT2, IFN regulatory factor 1 and 7) and a variety of IFN-inducible genes, including chemokines such as CCL5 and CXCL10. This result was consistent with our previous findings that showed significant downregulation of IFN-γ and several IFN-γ-inducible chemokines such as CXCL9 and CXCL10 within the CCR7-B16 tumors.4 The second group of genes consisted of MHC I and MHC II molecules, including beta-2-microglobin. The last group of downregulated genes included Tap1, Tap2 and tapasin. These proteins have well-characterized roles in endogenous peptide loading onto MHC I molecules of antigen-presenting cells and in the subsequent presentation of antigens to reactive CD4+ and CD8+ T cells.9, 10 Downregulation of MHC I surface expression and the resulting decreased efficiency of tumor antigen presentation to reactive T cells may explain the observed reduction of host anti-tumor immune response in popliteal DLNs of CCR7-B16-injected mice. Beta-2-microglobin as well as Tap1, Tap2 and tapasin proteins are all induced by type I and type II IFNs.11 Collectively, the characteristic deficiency of IFN-γ induction within CCR7-B16 tumors suggests that CCR7 overexpression leads to an apparent defect in tumor antigen presentation, and possibly, tumor antigen-specific T-cell priming.
Next, we focused on potential mediators of the IFN receptor signaling, including the IFN receptor itself, JAK/STAT transcription factors and other known STAT proteins. No significant difference in levels of IFN receptor mRNA was detected in CCR7-B16 versus pLNCX2-B16 tumor environment (Figure 2a). Similarly, the four JAK family members examined (JAK1, JAK2, JAK3 and Tyk2) were found to be expressed at similar levels in both tumor types (Figure 2b). However, STAT1, and to a lesser degree, STAT2 and STAT3, was strikingly downregulated in CCR7-B16 tumors relative to control tumors, whereas there was no significant difference in STAT4, STAT5a, STAT5b and STAT6. These findings suggest that tumoral CCR7 downregulates expression of specific STAT proteins relevant to IFN signaling, whereas cytokines and growth factors specific to other signaling pathways remain unaffected (Figure 2b).

Expression of IFN receptor, STATs and JAK family members in B16 tumors. Gene expression was analyzed by Affymetrix microarrays in three independent samples of CCR7-B16 and pLNCX2-B16 mouse tumors after CD45+ cell depletion. Gene expression levels of IFN-γ receptors (a) and of IFN-γ signal pathway genes (b) from Affymetry microarray-derived data are presented. Microarray results were validated using quantitative real-time PCR. The mRNA expression of 29 genes (Supplementary Table 2) was measured by quantitative real-time PCR analysis in tumor samples before and after CD45+ cell depletion and in in-vitro-cultured CCR7-B16 and pLNCX2-B16 cells. Relative expression levels of representative genes are shown for each of three groups: genes involved in the IFN-γ signaling pathways (STAT1, STAT2, STAT3, IRF1), MHC I and MHC II molecules (B2m) and genes involved in antigen presentation for MHC I (Tapbp; c). *P<0.05; **P<0.01.
Validation of microarray results by quantitative real-time PCR
To validate microarray results, we designed primers for 29 genes of interest (Supplementary Table 2) and performed quantitative real-time PCR. We compared the gene expression levels of the in-vitro-cultured B16 cell lines (CCR7-B16 and pLNCX2-B16) with their mouse footpad tumors before and after CD45 depletion. Overall, we were able to confirm the differential expression of all 29 tested genes between CCR7-B16 tumors and pLNCX2-B16 tumors validating the microarray results (Figure 2c; data not shown). The majority of genes tested were expressed at similar levels in CCR7-B16 and pLNCX2-B16 cell lines in vitro before inoculation in mice. We were able to independently confirm the downregulation of representative genes in each of the three gene groups described above. STAT5a and STAT5b serve as controls to show the specific downregulation of STAT1 in CCR7-B16 tumors (Supplementary Figure 1). These findings suggest that the downregulated genes in CCR7-B16 tumors are mainly of tumor and stromal origin (as opposed to products of tumor-infiltrating immune cells), as expression patterns were not significantly changed after CD45 depletion.
STAT1 proteins are downregulated in CCR7-B16 tumors compared with pLNCX2-B16 tumors
To confirm that STAT1 was downregulated at the protein level in CCR7-B16 tumors, we adopted two approaches. By western blot, we compared tumor samples before and after CD45 depletion. Total STAT1 protein was significantly reduced in CCR7-B16 tumors compared with pLNCX2-B16 tumors, regardless of whether tumor-infiltrating leukocytes were depleted (Figure 3a and Supplementary Figure 2). Flow cytometry analysis of STAT1 expression also detected decreased STAT1 expression in CCR7-B16 cells versus controls after gating on the CD45-negative cell population. On the basis of mean fluorescence intensity, pLNCX2-B16 tumors, on average, expressed three times more STAT1 protein than CCR7-B16 tumors (66.27±5.06 versus 23.23±7.07; P=0.001).

STAT1 expression and CCR7-B16 tumorigenesis. STAT1 protein levels in tumor samples before and after CD45+ cell depletion were evaluated by western blot. Cropped STAT1 and β-actin bands are shown, but a full-length blot is presented in Supplementary Figure 2A (a). CCR7-B16 cells were incubated overnight in the presence or absence of mouse IFN-γ (mIFN-γ; 1 ng/ml) before mouse footpad inoculation. Flow cytometry analysis for STAT1 expression in IFN-γ-treated and non-treated CCR7-B16 cells is shown, with mean fluorescent intensities of 97.75 and 14.6 (b). After inoculation of IFN-γ-treated and non-treated CCR7-B16 cells into the footpads, resulting footpad tumor volumes (mm3) were measured by a caliper on day 24 (c).
STAT1 upregulation by IFN-γ treatment in pLNCX2-B16 and CCR7-B16 cell lines in vitro is not prevented by CCR7 ligands
Next we determined if prior activation of CCR7 by its cognate ligand would prevent B16 cells from expressing STAT1 in vitro. We first evaluated the responsiveness of B16 cell lines to IFN-γ. Both pLNCX2-B16 and CCR7-B16 cells were treated with varying concentrations of mouse IFN-γ for 24 h and were then analyzed for STAT1 expression via flow cytometry. We found that STAT1 reached maximum expression levels when cells were treated with 250 pg/ml of mouse IFN-γ (data not shown). Both pLNCX2-B16 and CCR7-B16 cells were found to have similar levels of STAT1 in the absence of mouse IFN-γ treatment (Supplementary Figure 3). Both cell lines responded rapidly to IFN-γ treatment showing similar robust STAT1 induction (Supplementary Figure 3). To test whether or not activation of CCR7 by its ligand could abrogate the upregulation of STAT1 by IFN-γ, we pretreated both B16 cell lines with mCCL21 or hCCL19-Ig under different conditions (soluble versus immobilized) for varying time periods (6, 24 or 48 h) before adding 250 pg/ml of IFN-γ. The CCR7 activation did not prevent STAT1 activation in CCR7-B16 cells (Supplementary Figure 1), indicating that the downregulation of IFN-γ-mediated pathways in the CCR7-B16 tumor environment is most likely the result of the lack of IFN-γ production (as opposed to the ability of CCR7 activation to impede IFN-γ-mediated signaling).
IFN-γ-treated CCR7-B16 cells form smaller tumors in vivo
We hypothesized that CCR7-B16 tumors inhibited host anti-tumor immune response by downregulating IFN-γ production in vivo through unknown mechanisms. To test this hypothesis, we asked if transient upregulation of STAT1 expression in CCR7-B16 through IFN-γ stimulation before implantation could influence tumorigenesis. We incubated CCR7-B16 cells with or without 1 ng/ml mouse IFN-γ overnight before inoculating those cells into footpads of B6 wild-type mice. The upregulation of STAT1 in IFN-γ-treated CCR7-B16 cells was confirmed before injection by flow cytometry (Figure 3b). As IFN-γ may induce apoptosis in certain tumor cell lines, we also examined the apoptosis status by Annexin-V staining. Less than 1% of treated and untreated CCR7-B16 cells underwent apoptosis immediately before inoculation (data not shown). After inoculation into mouse footpads, IFN-γ-treated CCR7-B16 cells developed significantly (P=0.0013) smaller tumors by day 24 (Figure 3c). These experiments show that IFN-γ-induced activation of the STAT1 pathway affects CCR7-B16 tumorigenesis, suggesting a key role for this pathway in CCR7-B16 tumor immunity.
CCR7-B16 cells induce intratumoral lymphatic vessel formation
Affymetrix microarray analysis revealed that mRNA for podoplanin, a marker for lymphatic endothelium,12 was expressed at 3.35-fold higher levels in CCR7-B16 tumors than in pLNCX2-B16 tumors (Supplementary Table 1), suggesting lymphatic vessels were more abundant in CCR7-B16 tumors. To confirm this finding, we harvested CCR7-B16 and control tumors, and stained them with podoplanin-specific monoclonal antibody (mAb) 20 days after inoculation into ear skin (Figure 4a; see Supplementary Figure 7 for isotype control staining of mAbs against podoplanin, VEGF-C and CCL21). Podoplanin+ vessels were more abundant within CCR7-B16 tumors compared with control tumors (Figure 4a). Specifically, an increased density of podoplanin+ vessels was noted both within the central and peripheral areas of CCR7-B16 tumors. Even within a smaller CCR7-B16 tumor, abundant podoplanin-positive vessels were observed (Figure 4a, right panel), suggesting increased lymphatic vessel formation was not simply a function of tumor size.

Podoplanin and VEGF-C staining in CCR7-B16 tumors. CCR7-B16 or pLNCX2-B16 tumors were harvested 20 days after tumor cell inoculation into ears of mice. Anti-podoplanin antibody (green; a, b) and anti-VEGF-C antibody (red; b) were used for immunofluorescence and immunohistochemical staining (for quantification of lymphatic vessels). Four random high-power fields (HPF), which contained lymphatic vessels (podoplanin-positive structures with vessel morphology) or VEGF-C-expressing cells in each section, were selected. The numbers of the lymphatic vessels and of the VEGF-C-expressing cells were counted, and representative results of two tumor samples are shown. Two sections were stained from each tumor for VEGF-C (b). CCR7-B16 or pLNCX2-B16 tumors were harvested 20 days after tumor cell inoculation into footpads of C57BL6 mice. Equal-sized tumors were collected and homogenized. CD45+ cells were sorted from tumor suspensions using FACSAria II and were attached to a slide glass using Shandon Cytospin 3 Centrifuge. Sections were stained using anti-VEGF-C antibody. Three HPFs, which contained VEGF-C-expressing cells, in each section were selected and VEGF-C-expressing cells were counted (c). Scale bars, 100 μm.
Others have identified significant VEGF-C mRNA upregulation in human LN that contained nodal melanoma metastases, suggesting VEGF-C expression might identify tumors with a high risk for nodal metastases.13 To determine if VEGF-C expression was altered in CCR7-B16 tumors, we stained CCR7-B16 tumors using anti-VEGF-C antibody and anti-podoplanin antibody, and quantified VEGF-C-expressing cells within the CCR7-B16 tumors (Figure 4b, Supplementary Figure 4A). CCR7-B16 tumors showed approximately two-fold greater numbers of VEGF-C+ cells compared with control tumors (Figure 4b). Our microarray analysis did not detect upregulation of VEGF-C mRNA in CD45+ cell-depleted CCR7-B16 tumors (Supplementary Table 1), suggesting CD45+ cells might be the source of VEGF-C. Therefore, we isolated CD45+ cells from equal-sized tumors using FACSAria II (Supplementary Figure 4B), adhered them to glass slides for VEGF-C staining, and found an increased number of VEGF-C-expressing CD45+ cells in CCR7-B16 tumors compared with pLNCX2-B16 tumors (Figure 4c). These findings suggest that CCR7-B16 cells are capable of inducing lymphangiogenesis (despite the relative lack of inflammatory cells within tumors), and that this process is potentially mediated by upregulation of VEGF-C in CD45+ cells within the CCR7-B16 tumor microenvironment.
CCR7-B16 cells invade lymphatic vessels more efficiently than control B16 cells
CCL21 is a known ligand of CCR7 and functions as a chemoattractant for both malignant and non-malignant CCR7+ cells.14, 15 In mice, CCR7-B16 have been shown to metastasize to DLN with increased frequency when compared with control B16 cells.3 To determine whether CCR7-B16 cells invaded lymphatic vessels more efficiently, we first induced CCR7-B16 and control B16 tumors of similar size by inoculating non-limiting numbers of tumor cells into the footpads of mice as previously described.4 We hypothesized that CCL21, which is expressed on lymphatic endothelium, has a key role in facilitating the invasion of lymphatic vessels by CCR7-B16 cells. As shown in Supplementary Figure 5A, podoplanin-positive lymphatic vessels were present within CCR7-B16 tumors (left panel), and CCR7-B16 cells (as detected by a mAb cocktail against melanocyte/melanoma antigens) co-localized within or around these structures (Supplementary Figure 5A, right panel; Supplementary Figure 5B, isotype staining comparison in).
Quantification of both lymphatic vessel number and the number of intraluminal tumor cells revealed that both values were significantly increased in CCR7-B16 cell footpad tumors compared with controls (Figure 4a; Figure 5, left panel). Interestingly, there were also significantly greater numbers of intraluminal tumor cells per lymphatic vessel within CCR7-B16 tumors ( Figure 5, right panel), suggesting CCR7-B16 cells more efficiently invaded lymphatic vessels than did control cells. CCL21 staining was present in both intratumoral as well as peritumoral areas in close approximation with podoplanin+ vessels (Supplementary Figure 6). The findings above suggest that CCR7-B16 cells may be attracted to the increased number of lymphatic vessels via CCL21, resulting in increased transendothelial migration and subsequent nodal metastasis.

Invasion of CCR7-B16 cells into CCL21+ lymphatic vessels. C57BL6 mice footpad CCR7-B16 tumors were harvested 20 days after inoculation. To statistically assess the difference between lymphatic invasion in CCR7-B16 and control tumors, we stained sections of mouse ear tumors for podoplanin by immunohistochemical methods at day 20 and then counterstained the sections with hematoxylin. Four high-power fields (HPF), which contained the apparent lymphatic vessels in each section, were selected for analysis. The numbers of the lymphatic vessels and tumor cells (identified by hematoxylin staining) that invaded into the lymphatic vessels were counted (See Materials and Methods). Representative results of two tumor samples are shown.
Discussion
Herein, we demonstrated CCR7 expression by B16 cells prevented the induction of IFN-γ, thus blocking multiple anti-tumor pathways. Moreover, CCR7-B16 cells was highly associated with increased tumor lymphangiogenesis. We believe that the increased lymphangiogenesis observed in CCR7-B16 tumors is surprising and furthers our understanding of CCR7 in LN metastasis. Previously, our knowledge of the role of CCR7 in LN metastasis was primarily restricted to chemotaxis via expression of CCL21 by lymphatic vasculature and secondary lymphatic organs. In our studies, an increased number of lymphatic vessels were seen in CCR7-B16 tumors relative to control B16 tumors. These lymphatic vessels expressed CCL21, which would enable CCR7-B16 cells to invade them with increased efficiency relative to B16 cells. In support of this possibility, CCR7-B16 cells were present within podoplanin-positive lymphatic vessels in significantly greater numbers than control cells.
The observation that CCR7-B16 more effectively metastasized to regional LNs and formed primary tumors (when limiting numbers of tumor cells were inoculated) led us to investigate the underlying molecular mechanisms enabling escape from tumoral immunosurveillance. We found that fewer IFN-γ-expressing T cells were present in CCR7-B16 tumors. Further supporting this finding, STAT1/2, molecules specific to signaling events downstream of IFN-γ receptor engagement, were downregulated. In addition, tumor antigen presentation may also be affected, as the MHC I and MHC II genes were downregulated in the tumor microenvironment, which possessed markedly fewer IFN-γ-producing T cells. Collectively, this study suggests CCR7 expression on B16 cells promotes tumorigenesis in two ways: 1) immune evasion via the downregulation of IFN-γ and IFN-γ response genes, and 2) the induction of lymphangiogenesis with subsequent lymphatic invasion and nodal metastasis.
Recent clinical and experimental evidence have helped to define the numerous pathways, processes and gene products that regulate nodal metastasis. These include hypoxia, low pH, inflammatory mediators such as tumor necrosis factor-α,16, 17 chemokines and chemokine receptors like CCR7 and CCR10,18 epithelial to mesenchymal transition19 and lymphangiogenesis.20 Clinical evidence suggests the extent of lymphangiogenesis in primary cutaneous melanoma predicts the presence of sentinel LN metastases at the time of surgery and is therefore a novel prognostic indicator.20, 21 In a retrospective study, increased melanoma-associated lymphangiogenesis is inversely correlated with both disease-free survival and overall patient survival.22 Others have shown that, in vitro, metastatic melanoma expressing CCR7 have increased migration toward CCL21 containing conditioned medium from lymphatic endothelial cells.23 Our findings add to these observations by showing CCR7-expressing melanoma cells increase lymphatic density within (and surrounding) the tumor environment, thus providing avenues for the CCR7-expressing B16 cells to localize to these new, CCL21-positive lymphatic vessels.
In summary, we show CCR7 expression by melanoma may indicate an aggressive phenotype with enhanced nodal metastasis that is mediated by immune evasion and the promotion of lymphangiogenesis. To the best of our knowledge, our studies are the first to suggest that CCR7 expression may trigger enhanced lymphangiogenesis, possibly through a VEGF-C-mediated mechanism. Furthermore, our data suggests that CCR7-expressing melanoma cells escape immune surveillance early in tumorigenesis. Increased podoplanin expression by CCR7+ melanoma tumors may pose as a marker of poor prognosis and warrants further investigation.
Materials and methods
Animals and cell lines
Female C57BL/6 mice (8–12 weeks old) were purchased from the NCI Animal Production Colony (Frederick, MD, USA) or The Jackson Laboratory (Bar Harbor, ME, USA), and used in accordance with the guidelines of the Animal Use and Care Committee of NCI and the Medical College of Wisconsin. Syngeneic B16/F1 melanoma cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) with 10% heat-inactivated fetal bovine serum and supplements as previously described.3
Retroviral transduction of B16/F1 melanoma cells
B16/F1 melanoma cells were retrovirally transduced with mCCR7 cDNA (CCR7-B16 cells) or with empty vector (pLNCX2-B16 cells) using a pLNCX2 vector (Clontech, Mountain View, CA, USA) as previously described.4
Subcutaneous inoculation of transduced cell lines
The cell lines described above were harvested in exponential growth phase by trypsinization and washed in phosphate-buffered saline (PBS) before injection. For footpad injections, B16 cells (4 × 105 in 20 μl PBS) were injected into the left hind footpad. For ear injections, B16 cells (1 × 105 in 20 μl PBS) were injected into the subcutaneous space under the central dorsal surface of the ears immediately above the cartilage. Tumor growth was measured with a caliper and approximated by multiplying maximal tumor depth, width and perpendicular length.
ELISPOT assay for IFN-γ-producing cells in popliteal DLNs
ELISPOT reagents for detecting murine IFN-γ were purchased from BD Bioscience (San Diego, CA, USA). For ELISPOT assays, pLNCX2-B16 or CCR7-B16 cells (4 × 105 per injection) were inoculated into footpads of C57BL/6 wild-type mice. Popliteal DLNs from injected footpads and contralateral popliteal LNs from uninjected footpads were harvested at predefined time points. LNs2, 3 from the same group were randomly pooled together and considered as one sample. LN cells (1 × 106) were stimulated with 104 corresponding B16 cells in a pre-coated ELISPOT plate (Millipore, Bedford, MA, USA) for 24 h and counted with an ELISPOT analyzer (CTL, Cleveland, OH, USA).
Sample preparation for Affymetrix microarray analysis
CCR7-B16 cells or pLNCX2-B16 cells (4 × 105) were inoculated into the left hind footpad of C57BL/6 wild-type mice. Three weeks later, tumors were excised, minced and incubated in B16 culture medium containing 1 mg/ml of collagenase D (Roche, Indianapolis, IN, USA) and 0.5 mg/ml of DNase I (Sigma, St Louis, OH, USA) at 37 °C for 45 min. Tumor pieces were then gently disrupted using the back of a syringe plunger and filtered through a 40-μm spleen filter (BD Bioscience). Cells (1 × 106) from single-cell suspensions of pLNCX2-B16 or CCR7-B16 footpad tumors were mixed with 10 μl of anti-CD45 magnetic beads (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s protocol, washed and then depleted of CD45+ cells by placement through a magnetic separation column. Depletion of CD45-positive cells (>99%) was confirmed by fluorescence-activated cell sorting staining with fluorescein isothiocyanate-anti CD45 antibody. For Affymetrix microarray analysis (Affymetrix, Santa Clara, CA, USA), three independent sets of CD45+-depleted tumor samples were prepared and total RNA were extracted using RNeasy kit (QIAGEN, Valencia, CA, USA). Hybridizations to Affymetrix Mouse genome 430 2.0 Arrays were performed by NIDDK microarray core facility and the results were analyzed by ChipInspector and BiblioSphere software (Genomatix, Munich, Germany).
Quantitative real-time PCR to confirm microarray results
Single-cell suspensions of pLNCX2-B16 or CCR7-B16 footpad tumors collected before and after CD45+ cell depletion were prepared as described above. CCR7-B16 and pLNCX2-B16 cells cultured in vitro in regular B16 medium were harvested and washed in PBS. Total RNA was extracted from tumor cells or in-vitro-cultured cell lines using RNeasy kit (QIAGEN). Total RNA (3 μg) was converted into cDNA using SuperScript II First-strand synthesis kit (Invitrogen). Quantitative reverse-transcriptase PCR primers were designed using Genscript (www.genscript.com) online program, and primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). SYBR Green PCR master mix was purchased from Applied Biosystems (Carlsbad, CA, USA) and real-time PCR was performed in a Chromo4 real-time PCR machine (Bio-Rad, Hercules, CA, USA). Gene expression levels were normalized to mouse glyceraldehyde 3-phosphate dehydrogenase. Primer pairs are shown in Supplementary Table 1. Gene expression by quantitative reverse-transcriptase PCR was measured in two independent samples different from the samples analyzed in Affymetrix microarray. The Student’s t-test was used to assess significance.
Western blotting and flow cytometry to detect STAT1 proteins
Single-cell suspensions of pLNCX2-B16 or CCR7-B16 footpad tumors collected before and after CD45+ cell depletion were prepared as described above. Total proteins were extracted using RIPA buffer (Sigma) containing protease inhibitor cocktail (Roche). After centrifugation at 14 000 r.p.m. for 15 min at 4 °C, supernatants were collected and protein concentration was determined using RC DC protein assay kit (Bio-Rad). Cell lysates (50 μg total protein) were denatured, loaded onto 10% SDS–polyacrylamide gel electrophoresis (Invitrogen) and transferred to nitrocellulose membrane (Invitrogen). Membranes were blocked with 5% blotting grade non-fat dry milk (Bio-Rad) for 1 h and incubated with anti-STAT1 mAb (BD Bioscience) at 1:500 dilutions for 1 h. Membranes were then washed three times and incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (Promega, Madison, WI, USA) at 1:2500 dilutions for 1 h. After washing, membranes were incubated with chemiluminescent solution (SuperSignal West Pico Substrate; Thermo Scientific, Rockford, IL, USA) for 5 min at room temperature (RT) and exposed to film. The same membrane was then stripped with stripping buffer (Thermo Scientific), followed by incubation with horseradish peroxidase-conjugated anti-β actin antibody (Sigma) at 1:50 000 dilution.
For flow cytometry analysis, single-cell suspensions of pLNCX2-B16 or CCR7-B16 footpad tumors were first stained with fluorescein isothiocyanate-CD45 at 4 °C for 30 min. After washing, tumor cells were fixed with 250 μl of fixation buffer (BD Bioscience) at 37 °C for 10 min, followed by permeabilization using Perm buffer III (BD Bioscience) on ice for 30 min. Tumor cells were then washed and subsequently stained with PE-STAT1 (BD Bioscience) for 30 min at 4 °C. Samples were then collected on FACSCalibur machine (BD Bioscience) and analyzed with FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Immunohistochemistry
We purchased hamster anti-podoplanin antibody (8.1.1) from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA, USA), anti-VEGF-C antibody (H-190) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and mouse CCL21/6Ckine Biotinylated Antibody from R&D Systems (Minneapolis, MN, USA). For melanosome staining, we purchased a premixed mAb cocktail from Abcam (Cambridge, MA, USA; mouse monoclonal (HMB45+DT101+BC199) to melanoma (ab732)). As secondary antibodies for imunofluorescence, we purchased Alexa Fluor 488 goat anti-hamster IgG (H+L), Alexa Fluor 568 goat anti-mouse IgG (H+L), and tyramide signal amplification kit #24 from Invitrogen, and DyLight 594 AffiniPure Goat Anti-Rat IgG (H+L) from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
B16 ear tumors were excised, embedded in optimal cutting temperature compound without fixation, frozen and then sectioned. CD45+ cells were isolated from cell suspensions of pLNCX2-B16 or CCR7-B16 footpad tumors using FACSAria II (BD Bioscience) and were attached to the slide glasses using Shandon Cytospin 3 Centrifuge (Thermo Scientific). Tissue sections were fixed with either ice-cold acetone for 10 min or with paraformaldehyde solution, 4% in PBS (USB Corp., Cleveland, OH, USA) for 2 h, followed with 95% ethanol for 20 min. Fixed sections were blocked for 1 h at RT with 5% goat serum and Fc-blocker (2.4G2, Bio X Cell, West Lebanon, NH, USA) in PBS containing 3% skim milk, and incubated with primary antibody overnight at 4 °C. For HMB45, DT101 and BC199 staining, tissue sections were fixed with 4% paraformaldehyde PBS solution (USB Corp.) for 10 min, then incubated in PBS containing 0.5% Triton X-100 for 5 min. Samples were blocked for 1 h at RT with 5% goat serum, Fc-blocker (2.4G2, Bio X Cell) and donkey anti-mouse IgG-horseradish peroxidase (Santa Cruz) in PBS containing 3% skim milk, and incubated with primary antibody overnight at 4 °C. For immunofluorescence analysis, sections were fixed, incubated with blocking buffer and primary antibody as described above. The sections were then washed with PBS, incubated with secondary antibody for 30 min at RT or stained using Tyramide Signal Amplification Kit number 24 according to the manufacturer’s protocol. After staining, cells were mounted with SlowFade Gold Antifade Reagent with 4',6-diamidino-2-phenylindole (Invitrogen). Images were acquired using a Carl Zeiss microscope (Carl Zeiss, Oberkochen, Germany). For counting lymphatic vessels and melanoma cells invading into them, sections were blocked and incubated with anti-podoplanin antibody as described above. The sections were then washed with PBS and incubated with biotinylated anti-hamster IgG (H+L; Vector Laboratories Inc., Burlingame, CA, USA) for 30 min at RT, stained with an avidin–biotin method using DAB Peroxidase Substrate Kit as a substrate for peroxidase (VECTASTAIN Elite ABC Kit (Standard*) from Vector Laboratories Inc.) and counterstained with hematoxylin. Images were acquired using a Carl Zeiss microscope, and the numbers of the lymphatic vessels (podoplanin+ structures with vessel morphology) and of tumor cells (identified as medium- to large-sized cells with large nuclei by hematoxylin staining) that were clearly within or closely associated with (indicating invasion) lymphatic vessels were counted.
Statistical analysis
A paired two-tailed Student’s t-test was used to analyze the results and a P-value<0.05 was considered statistically significant. All the shown values represent means and s.e.m.
References
Fang L, Hwang ST Roles of CCR7 in Cancer Biology. In: Fulton AM (ed.) Chemokine Receptors in Cancer. Humana Press, New York, 2009, pp 93–108.
Mashino K, Sadanaga N, Yamaguchi H, Tanaka F, Ohta M, Shibuta K et al. Expression of chemokine receptor CCR7 is associated with lymph node metastasis of gastric carcinoma. Cancer Res, 2002 62: 2937–2941.
Wiley HE, Gonzalez EB, Maki W, Wu MT, Hwang ST Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J Natl Cancer Inst, 2001 93: 1638–1643.
Fang L, Lee VC, Cha E, Zhang H, Hwang ST CCR7 regulates B16 murine melanoma cell tumorigenesis in skin. J Leukoc Biol, 2008 84: 965–972.
Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci USA, 1998 95: 258–263.
Saeki H, Moore AM, Brown MJ, Hwang ST Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol, 1999 162: 2472–2475.
Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G et al. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med, 2001 194: 797–808.
Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med, 2001 7: 192–198.
Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity, 1996 5: 103–114.
Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4-8+ T cells. Cell, 1992 71: 1205–1214.
Ramadori G, Mitsch A, Rieder H, Meyer zum Buschenfelde KH Alpha- and gamma-interferon (IFN alpha, IFN gamma) but not interleukin-1 (IL-1) modulate synthesis and secretion of beta 2-microglobulin by hepatocytes. Eur J Clin Invest, 1988 18: 343–351.
Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E et al. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol, 1999 154: 385–394.
Goydos JS, Gorski DH Vascular endothelial growth factor C mRNA expression correlates with stage of progression in patients with melanoma. Clin Cancer Res, 2003 9: 5962–5967.
Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res, 2009 69: 349–357.
Schweickart VL, Raport CJ, Godiska R, Byers MG, Eddy RL, Shows TB et al. Cloning of human and mouse EBI1, a lymphoid-specific G-protein-coupled receptor encoded on human chromosome 17q12-q21.2. Genomics, 1994 23: 643–650.
Bedogni B, Powell MB Hypoxia, melanocytes and melanoma - survival and tumor development in the permissive microenvironment of the skin. Pigment Cell Melanoma Res, 2009 22: 166–174.
Zhu N, Lalla R, Eves P, Brown TL, King A, Kemp EH et al. Melanoma cell migration is upregulated by tumour necrosis factor-alpha and suppressed by alpha-melanocyte-stimulating hormone. Br J Cancer, 2004 90: 1457–1463.
Kakinuma T, Hwang ST Chemokines chemokine receptors, and cancer metastasis. J Leukoc Biol, 2006 79: 639–651.
Lee JM, Dedhar S, Kalluri R, Thompson EW The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol, 2006 172: 973–981.
Dadras SS, Lange-Asschenfeldt B, Velasco P, Nguyen L, Vora A, Muzikansky A et al. Tumor lymphangiogenesis predicts melanoma metastasis to sentinel lymph nodes. Mod Pathol, 2005 18: 1232–1242.
Massi D, Puig S, Franchi A, Malvehy J, Vidal-Sicart S, Gonzalez-Cao M et al. Tumour lymphangiogenesis is a possible predictor of sentinel lymph node status in cutaneous melanoma: a case-control study. J Clin Pathol, 2006 59: 166–173.
Dadras SS, Paul T, Bertoncini J, Brown LF, Muzikansky A, Jackson DG et al. Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol, 2003 162: 1951–1960.
Shields JD, Emmett MS, Dunn DB, Joory KD, Sage LM, Rigby H et al. Chemokine-mediated migration of melanoma cells towards lymphatics--a mechanism contributing to metastasis. Oncogene, 2007 26: 2997–3005.
Yamazaki F, Okamoto H, Matsumura Y, Tanaka K, Kunisada T, Horio T Development of a new mouse model (xeroderma pigmentosum a-deficient, stem cell factor-transgenic) of ultraviolet B-induced melanoma. J Invest Dermatol, 2005 125: 521–525.
Acknowledgements
We thank Dr Ryan E Sells, Dr Xuesong Wu, Mr Nathan Duncan and Dr Tomotaka Mabuchi (Department of Dermatology, MCW) for their advice and technical support. This study was supported by the Advancing Healthier Wisconsin Research Funds, Advancing Healthier Wisconsin Endowment of the Medical College of Wisconsin and The Ann’s Hope Foundation for Melanoma (to STH), an MCW/Froedtert Cancer Center Collaborative Research Fellowship (to TT) and the Intramural Research Program of the National Cancer Institute (STH and LF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogenesis website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Takekoshi, T., Fang, L., Paragh, G. et al. CCR7-expressing B16 melanoma cells downregulate interferon-γ-mediated inflammation and increase lymphangiogenesis in the tumor microenvironment. Oncogenesis 1, e9 (2012). https://doi.org/10.1038/oncsis.2012.9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2012.9
Keywords
This article is cited by
-
CCL21-CCR7 signaling promotes microglia/macrophage recruitment and chemotherapy resistance in glioblastoma
Cellular and Molecular Life Sciences (2023)
-
Role of VEGF-A/C and CCR-7 in the enhanced metastasis of A549 cells induced by 2 and 4 Gy X-rays in vitro and in vivo
Science China Technological Sciences (2014)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}