
Abstract
Transformation by Simian Virus 40 (SV40) large T antigen (LT) is mediated in large part by its interaction with a variety of cellular proteins at distinct binding domains within LT. While the interaction of LT’s N-terminus with the tumor suppressor Rb is absolutely required for LT-dependent transformation, the requirement for the interaction of LT’s C-terminus with p53 is less clear and cell- and context-dependent. Here, we report a line of transgenic mice expressing a doxycycline-inducible liver-specific viral transcript that produces abundant 17kT, a naturally occurring SV40 early product that is co-linear with LT for the first 131 amino acids and that binds to Rb, but not p53. Comparative analysis of livers of transgenic mice expressing either 17kT or full length LT demonstrates that 17kT stimulates cell proliferation and induces hepatic hyperplasia but is incapable of inducing hepatic dysplasia or promoting hepatocarcinogenesis. Gene expression profiling demonstrates that 17kT and LT invoke a set of shared molecular signatures consistent with the action of LT’s N-terminus on Rb-E2F-mediated control of hepatocyte transcription. However, 17kT also induces a unique set of genes, many of which are known transcriptional targets of p53, while LT actively suppresses them. LT also uniquely deregulates the expression of a subset of genes within the imprinted network and rapidly re-programs hepatocyte gene expression to a more fetal-like state. Finally, we provide evidence that the LT/p53 complex provides a gain-of-function for LT-dependent transformation in the liver, and confirm the absolute requirement for LT’s C-terminus for liver tumor development by demonstrating that phosphatase and tensin homolog (PTEN)-deficiency readily cooperates with LT, but not 17kT, for tumorigenesis. These results confirm independent and inter-dependent functions for LT’s N- and C-terminus and emphasize differences in the requirements for LT’s C-terminus in cell-type dependent transformation.
Similar content being viewed by others


Introduction
Simian Virus 40 (SV40) large and small T-antigens (LT and st) transform cells by binding to a multitude of cellular proteins, most notably the tumor suppressors pRb and p53 and protein phosphatase 2A.1, 2 LT and st are produced from an alternatively spliced early viral pre-RNA in which the ∼2.2 kb LT and ∼2.5 kb st transcripts are generated by removal of a single intron between nucleotides 4918-4571 or nucleotides 4642-4571, respectively.3 For decades, LT and st were considered to be the only two early region products; however a third protein, 17kT, was subsequently found to be co-expressed with LT and st in productively infected cells.4 The 17kT is translated from a ∼1.5 kb transcript generated by removal of a second 746 bp intron from the LT transcript, resulting in a frame shift that generates a protein that is co-linear with LT for its N-terminal 131 amino acids with an additional four unique amino acids at its C-terminus. Given its co-linearity with LT, 17kT shares several regulatory domains with LT that mediate some of its most important functions including inactivation of Rb5 and override of the Bub1-induced spindle checkpoint,6 and as such, 17kT promotes cell cycle progression in the presence of st and complements LT J domain mutants that are defective for transformation in vitro.7 However, when compared with full length LT, 17kT is only weakly transforming for cultured cells,4 indicating that functions encoded downstream of amino acid 131 are required for LT’s full transforming activity.
In this study, we describe a unique line of transgenic mice expressing a conditional liver-specific transgene that produces 17kT and st in the absence of any detectable LT, and compare and contrast it with an equivalent line of transgenic mice that co-expresses LT, 17kT and st. Our results demonstrate that 17kT and LT are functionally equivalent at promoting hepatocyte proliferation and driving hepatic hyperplasia, yet only LT induces hepatic dysplasia and promotes hepatocarcinogenesis demonstrating that LT’s C-terminus is absolutely required for hepatocyte transformation. This is underscored by our demonstration that loss of the tumor suppressor phosphatase and tensin homolog (PTEN) fails to cooperate with 17kT for liver tumor development despite accelerating LT-induced tumorigenesis. By comparing and contrasting these two strains of mice, we identify molecular signatures associated with expression of 17kT and LT that provide insight into how LT’s N- and C-terminal domains regulate gene expression to specify proliferation or transformation in quiescent primary epithelial cells in vivo. We also provide evidence that LT’s C-terminus, most likely via formation of the LT/p53 complex, promotes transformation by partially re-programming the hepatic transcriptome.
Results
ApoE-rtTA:TRE2-TAg line 6-1 transgenic mice express 17kT and st, but not LT
Bi-genic ApoE-rtTA:TRE2-TAg transgenic mice expressing the 2.7 kb SV40 early region in the liver in a doxycycline (dox)-dependent manner were generated as described in Materials and methods and Supplementary Figure S1. Lines were established from founder mice 6-1 and 7-1, both of which exhibited Mendelian transmission of both transgenes. To assess dox-dependent hepatic expression of the SV40 transgene, wild-type (WT) and transgenic littermates from both lines were provided with drinking water supplemented with 10 or 25 μg/ml doxycycline (dox) for 4, 28 or 84 days. All treatment regimens resulted in hepatomegaly in transgenic mice of line 7-1; however only regimens of the longest durations induced liver growth in transgenic mice from line 6-1 (Figure 1A).

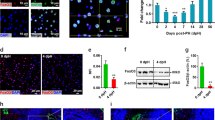
Livers of ApoE-rtTA:TRE2-TAg transgenic mice (line 6-1) express 17kT and st, but not LT. (A) Newly weaned WT and transgenic male mice from lines 6-1 and 7-1 were provided with 10 or 25 μg/ml dox in drinking water for 4, 28 or 84 days. Mice were killed and weighed and % liver/body weight values calculated. n⩾4 mice/group. Error bars +/− s.e.m. **P<0.05, ***P<0.001. (B) Immunoblot analysis of viral oncoprotein expression in livers of transgenic mice from line 6-1 and line 7-1. One-hundred microgram liver lysates prepared from WT (lane 1) and transgenic mice (lanes 2 and 3) provided with 25 μg/ml dox for 4 days were immunoblotted with SV40-specific antibodies UT450, pab101 and pab280. Lysates from a line of transgenic mice expressing a dox-inducible st transgene (ApoE-rtTA:TRE-st) (Comerford and Hammer, unpublished) (lane 4), and LT and st from the phosphoenolpyruvate carboxykinase (PEPCK) regulatory elements8 (lane 5) were used as controls for LT and st expression. (C) Northern blot analysis of total liver RNA from WT (lane 1), line 7-1 (lane 2) and 6-1 (lane 3) transgenic mice provided with 25 μg/ml dox for 4 days. The membrane was hybridized to a p32-dCTP (deoxycytidine triphopsphate)-labeled probe comprising the entire 2.7 kb SV40 early region. The blot was stripped and re-probed with an oligonucleotide specific for 18S rRNA to verify equal loading of RNA. (D) IHC (3′-amino-9-ethylcarbazole chromagen) of liver sections from transgenic mice of line 7-1 (a, c) and line 6-1 (b, d) with the LT-specific antibody pab101 without hematoxylin (a, b) or with hematoxylin counterstain (c, d). Magnification × 112.
To determine if the slower rate of liver growth in line 6-1 reflected a lower level of viral oncoprotein expression, we immunoblotted liver lysates prepared from dox-treated 6-1 and 7-1 transgenic mice, together with lysates from transgenic mice expressing LT and st from the phosphoenolpryruvate carboxykinase (PEPCK) promoter (PEPCK-TAg)8 and mice expressing st from a dox-inducible ApoE-rtTA:TRE2-st transgene (Comerford and Hammer, unpublished) with a panel of SV40-specific antibodies (Figure 1B). Antibody UT450, which recognizes LT and st,9 detected the 97 kDa LT protein in liver extracts of mice from line 7-1 and phosphoenolpyruvate carboxykinase (PEPCK)-TAg mice but not 6-1 mice, and the ∼19 kDa st protein in liver extracts of phosphoenolpyruvate carboxykinase (PEPCK)-TAg and ApoE-rtTA:TRE2-st mice. However, the only protein recognized by UT450 in 6-1 livers was an abundantly expressed protein (labeled *) that migrated at ∼17 kDa. Immunoblotting with pab101 and pab280, antibodies that recognize epitopes in the C-terminal 190 amino acids of LT or in st, respectively, confirmed the absence of LT in livers of 6-1 mice and expression of st in livers from all transgenic lines. Interestingly, despite the failure of UT450 to detect st in lysates from 6-1 or 7-1 mice, pab280 detected st in extracts from both, likely reflecting differences in protein/epitope conformation, antibody specificity, affinity or less abundant expression. Importantly, pab280 failed to recognize the ∼17 kDa protein in livers of 6-1 mice, suggesting that it was not a truncated st protein. Analysis with pab108, a monoclonal antibody that recognizes an epitope in LT’s N-terminus and st also recognized the ∼17 kDa protein in livers of line 6-1 (data not shown).
On the basis of antibody recognition and size, we suspected that livers of line 6-1 mice either expressed 17kT or a severely truncated LT protein. To distinguish between these two possibilities, we analyzed total liver RNA isolated from mice of each line by Northern blotting using the entire SV40 2.7 kb early region as probe, which confirmed expression of the expected 2.2 kb LT and the 2.5 kb st transcripts in livers from 7-1 mice (Figure 1C). However, an abundant transcript of ∼1.5 kb (labeled *) plus several larger, yet minor transcripts were evident in livers from 6-1 mice, none of which co-migrated with LT or st. Reverse transcription PCR (RT–PCR) of oligo-dT-primed cDNA generated from total liver RNA isolated from both the transgenic lines using a series of SV40 early-region-specific primers (Supplementary Figure S2 and Supplementary Table S2) confirmed the absence and presence of a full length LT transcript in livers of 6-1 and 7-1 mice, respectively, and expression of st in both the transgenic lines. RT–PCR also amplified a transcript of the expected size for 17kT in livers of 6-1 mice, which was subsequently confirmed as 17kT by direct sequencing of two independently generated amplicons (Supplementary Results; Supplementary Figure S3). Finally, immunohistochemistry (IHC) with pab101 confirmed hepatocyte-specific expression of LT in livers of 7-1, but not 6-1 mice (Figure 1D). Thus, while livers of transgenic mice from the 7-1 line expressed all three oncoproteins encoded by the SV40 early region, livers from line 6-1 only expressed 17kT and st. For simplicity, ApoE-rtTA:TRE2-TAg transgenic lines 6-1 and 7-1 are herein referred to as 17kT and LT, respectively.
17kT and LT stimulate hepatocyte proliferation, but only LT induces hepatic dysplasia and promotes hepatocarcinogenesis
To determine whether the differences in dox-dependent liver growth in each of the transgenic lines (Figure 1A) reflected differences in hepatocyte size, proliferation or survival, we performed histological and IHC analysis of livers from transgenic mice from both the lines without dox or provided with dox for 4 days or 4 weeks. Histological analysis showed that the rapid growth of LT livers after just 4 days of dox treatment predominantly reflected increased hepatocyte size due to the induction of LT-dependent dysplasia, which was absent from livers expressing 17kT (Supplementary Figure S4 and Supplementary Results). IHC for proliferating cell nuclear antigen (PCNA) (Supplementary Figure S4) and terminal deoxynucleotidyl transferase dUTP nick end labeling analysis (not shown) showed that LT and 17kT both forced hepatocytes back into the cell cycle without significantly increasing apoptosis. Sustained dox treatment for 4 weeks also promoted uniform hyperplastic liver growth in both the lines, reflecting ongoing hepatocyte proliferation in 17kT livers and the replacement of large dysplastic hepatocytes with smaller, highly proliferative hepatocytes in LT livers (Supplementary Figure S4). However, after ∼4–5 weeks of dox treatment, livers expressing LT began to exhibit multi-focal tumor development with 100% of mice exhibiting extensive tumor growth after ∼7.5 weeks. This, however, was in sharp contrast to livers expressing 17kT which remained hyperplastic and tumor-free, with only one mouse exhibiting tumor development before 14 weeks on dox (Figures 2A and B). Multi-focal hepatocellular carcinoma (HCC) also developed in 55% (6/11) of 24–30-week-old dox-naive 7-1 mice (data not shown), prompting us to analyze viral transcript expression in livers from dox-naive and dox-treated mice from both the lines by northern blotting, which confirmed that transgene expression was ‘leaky’ in both (Supplementary Figure S5). In contrast to 7-1 transgenic mice, however, leaky expression of 17kT had no significant effect on liver mass or hepatocyte morphology and failed to predispose to tumor development.

17kT is insufficient for HCC development. (A) Gross appearance of livers of WT (left) and ApoE-rtTA:TRE2-TAg transgenic mice expressing either 17kT (middle) or LT (right) provided with 25 μg/ml dox for 5.5 weeks. The 17kT liver is enlarged and uniformly hyperplastic while the LT liver is tumor-laden. (B) Kaplan–Meier curve of tumor-free incidence in livers of mice expressing 17kT and LT provided with 10 μg/ml dox. (C) IHC showing that tumors in livers of mice from line 6-1 develop from rare LT-expressing hepatocytes. IHC for LT in hyperplastic livers from mice from line 7-1 (a) and 6-1 (b) and from a tumor-bearing 6-1 mouse liver (dotted line defines nodule boundary) (c). Background staining in (b) and extra-nodular staining in (c) is evident due to non-specific secondary antibody staining as a consequence of extended length of incubation with chromagen (10 min vs 2 min), which was necessary to visualize the lower level of LT expressed in the nodule of the 17kT liver. 3′-amino-9-ethylcarbazole chromagen (orange/brown); no counterstain. Magnification × 112.
Although both transgenic lines had arisen from founders expressing viral pre-RNAs that differed in their ability to produce LT, we considered the possibility that the solitary tumors that developed in livers of 6-1 mice after long-term dox treatment might be due to the clonal expansion of rare LT-expressing cells. To determine if this was the case, we performed IHC with pab101 on sections of hyperplastic livers from 6-1 and 7-1 mice and on a solitary tumor-bearing 17kT-expressing liver (Figure 2C). While virtually all hepatocytes within the 7-1 liver demonstrated strong nuclear expression of LT (a), no LT was detected in hyperplastic 17kT liver (b. However, LT was expressed in nodular hepatocytes in the 17kT liver (c), albeit at a lower level than in the LT-expressing liver (a), confirming that rare LT-expressing hepatocytes had gained an oncogenic advantage and produced tumors within a background of 17kT-induced hyperplasia. Taken together, these results demonstrate that while 17kT is sufficient to drive hepatic hyperplasia, functions encoded in LT downstream of amino acid 131 are absolutely required for hepatocarcinogenesis.
LT, but not 17kT, re-establishes a fetal-like hepatic gene-expression program and alters the expression of multiple imprinted genes and growth regulators in adult liver
To gain insight into the molecular targets of 17kT and LT that could explain their differential tumor-inducing capabilities, we performed gene-expression array analysis of livers from WT and transgenic male mice from both the lines and compared their gene-expression profiles after inducing 17kT or LT with 25 μg/ml dox for 4 days, a regimen adopted to identify primary, rather than secondary targets of the viral proteins. Using a 3-fold cutoff for up- or downregulated genes, array analysis showed that despite the strikingly different hepatocyte morphologies induced by 17kT and LT after just 4 days of dox treatment, 635 of 1189 (53%) mRNAs were upregulated (Supplementary Table S3), and 379 of 915 (41%) mRNAs were downregulated by both the viral proteins (Supplementary Table S4), indicating that both viral proteins altered the expression of a common set of hepatic genes. mRNAs coordinately induced by 17kT and LT included those involved in DNA replication, mitotic regulation, cell cycle checkpoint monitoring and DNA repair, while those that were downregulated included mRNAs involved in lipid/lipoprotein metabolism, xenobiotic metabolism and hormone biosynthesis (Figure 3). Further analysis, however, revealed that many hepatic mRNAs also demonstrated differential regulation by each viral protein (Supplementary Table S3). For instance, LT preferentially induced the expression of mRNAs for histone genes, alpha-fetoprotein (afp), amy2, cd24a, prom1/CD133, h19, sox4, peg3, bex1, igf2 and c-Myc, while 17kT preferentially induced the expression of slitrk4, lmln, acot1, ak1 sulf2, ces5, psrc1/DDA3 and eda2r (Supplementary Table S3). Given the functional diversity of these mRNAs, however, bioinformatics analysis failed to assign the majority of these transcripts to distinct processes or pathways (Figure 3). Common or differential regulation by each of the viral proteins was subsequently confirmed by semi-quantitative RT–PCR for a subset of mRNAs preferentially induced by LT (Figures 4A and B), 17kT (Figure 4C) or 17kT and LT (Figure 4D) using primers and conditions listed in Supplementary Table S5.

Gene-expression microarray profiling of 17kT and LT function in hepatocytes. Venn diagrams illustrating common and differential regulation of hepatic genes by 17kT and LT (⩾ 3-fold up (left (green/yellow)) or down(right (red/yellow))-regulated genes). In each case, the intersection between circles represents mRNAs coordinately regulated by 17kT and LT, while the left and right circles in each box represent genes preferentially regulated by 17kT or LT, respectively (see Materials and Methods for details). Genes were assigned to biological functions and processes using Reactome Skypainter (www.reactome.org/cgi-bin/skypainter2).

Verification of altered hepatic gene expression in livers expressing 17kT or LT. Genes identified by array analysis as being altered in 17kT or LT livers were verified by semi-quantitative RT–PCR (A, C, D), Northern blotting (B) or IHC (E). Gene-specific oligonucleotides are listed in Supplementary Table S5. (A) RT–PCR analysis of developmentally regulated, imprinted and growth-related mRNAs preferentially induced by LT. (B) Northern analysis of total liver RNA using a p32-dCTP (deoxycytidine triphosphate)-labeled mouse c-Myc-specific probe, followed by a stripping and re-probing with an 18S rRNA oligonucleotide probe. (C) mRNAs preferentially induced by 17kT. (D) mRNAs coordinately induced by 17kT and LT. RT–PCR for cyclophilin A was used to verify equal input of cDNA into reactions. The same cyclophilin A panel is shown at the bottom of (A), (C) and (D). (E) IHC for α-fetoprotein (afp) of WT (a), 17kT (b) and LT livers (c) (3′-amino-9-ethylcarbazole chromogen (red) with hematoxylin counterstain (blue)), magnification × 112. Note isolated hepatocyte demonstrating strong afp immunoreactivity (inset, magnification × 224) in 17kT livers (b) and strong uniform afp immunoreactivity in LT livers (c). All analyses were conducted using livers of newly weaned male mice provided with 25 μg/ml dox for 4 days.
Induction of h19, igf2, peg3, prom1/CD133 and rex3 in LT-expressing livers indicated that LT’s C-terminus was preferentially altering the expression of mRNAs within the imprinted network and inducing HCC-associated markers.10, 11 Similarly, induction of mRNAs such as afp and cd24a suggested that LT was re-activating the expression of mRNAs highly expressed during fetal liver development12, 13, 14, 15, 16, 17 (Supplementary Table S6). To determine if LT was inducing the focal expansion of fetal hepatic stem cells that were not histologically discernible after just 4 days of dox treatment or was rapidly altering the differentiation status of the adult liver, we analyzed afp expression by IHC in livers of WT and transgenic mice from both the lines provided with dox for 4 days (Figure 4E). While no afp-positive cells were visible in WT livers, rare, solitary afp-positive hepatocytes could be seen in livers expressing 17kT (Inset). In contrast, the majority of the large dysplastic hepatocytes in LT-expressing livers demonstrated intense cytoplasmic afp staining, consistent with afp being the most highly induced mRNA in LT-expressing livers. IHC also confirmed that altered cell fate and differentiation correlated with LT-dependent tumor development after 5–6 weeks of dox treatment (Supplementary Figure S6; Supplementary Results). Taken together, these results show that while LT’s N-terminus is sufficient to drive hepatic hyperplasia, induction of hepatic dysplasia and tumor development require C-terminal function(s) that drive the re-programming of adult hepatocyte gene expression to a more fetal-like state.
LT’s C-terminus actively represses the expression of transcriptional targets induced by LT’s N-terminus and binds and stabilizes p53 specifically in tumors
Although Rb inactivation activates p53 to counteract unscheduled E2F-dependent proliferation,18, 19 17kT-expressing livers became hyperplastic, suggesting that 17kT either abrogated or circumvented the p53-dependent checkpoint. To determine if 17kT-expressing hepatocytes proliferated by preventing p53-dependent transcription or in spite of it, we compared mRNAs preferentially upregulated by 17kT with known p53 targets, interactors and with genes otherwise implicated in p53-dependent processes. As LT’s C-terminus also inhibits p53-dependent transcription by preventing p53 from binding to DNA,20, 21 we also interrogated the arrays to determine the extent to which LT suppressed any p53 targets. Supplementary Table S7 shows that of the 129 different mRNAs preferentially induced by 17kT, 26 (20%) have either been identified as bona fide p53 targets or are implicated in p53-dependent processes, demonstrating that 17kT promoted hepatocyte proliferation despite ongoing p53-dependent transcription. Moreover, LT actively suppressed or neutralized the expression of a subset of 17kT-induced mRNAs including slitrk4, lmln, ces5 and the p53 target, sulf2, implicating these as possible hepatic targets of LT-mediated repression of p53. Thus, while 17kT drives hyperplastic growth by disrupting Rb-E2F-dependent transcriptional control and overriding the p53-dependent checkpoint, LT’s C-terminus promotes hepatic dysplasia and likely predisposes to tumorigenesis by disrupting Rb-E2F, re-programming hepatic gene expression and dominantly repressing a subset of mRNAs induced by its N-terminus.
Evidence indicates that the LT/p53 complex provides a gain-of-function mechanism for LT-dependent transformation that is distinct from inactivation of p53-dependent transcription.22, 23 However, data from in vivo mouse models expressing truncated or full length LT also suggest that certain cell types do not require LT’s C-terminus or p53 binding for transformation.24, 25, 26, 27 To determine if p53 was stabilized and thus presumably required for LT-dependent liver tumorigenesis, we performed IHC on serial sections of tumor-bearing LT-expressing livers, hyperplastic 17kT-expressing livers and WT livers with LT- and p53-specific antibodies. Consistent with the short half-life of p53 in hepatocytes and the lack of LT expression in hyperplastic 17kT livers, neither p53 nor LT were detected in WT and 17kT livers (not shown). However, while LT was expressed in almost every hepatocyte in 7-1 livers (Figures 5a), it was most highly expressed in nests of hyperproliferating hepatocytes and tumor nodules. Interestingly, although p53 protein was also readily detectable in LT livers, it was most evident in nodular hepatocytes expressing the highest levels of LT (Figures 5b). The stabilization and co-localization of p53 and LT specifically in tumor nodules strongly suggests that both proteins interact and that the LT/p53 complex provides an oncogenic advantage for tumor development in the liver.

LT and p53 are specifically stabilized and co-localized in tumor nodules in livers of 7-1 mice. Five micrometers formalin-fixed paraffin-embedded serial sections from tumor-bearing LT-expressing livers of mice provided with 25 μg/ml dox for 5.5 weeks were subjected to immunohistochemical analysis for LT (a, c, e) and p53 (b, d, f). p53 is preferentially stabilized in nodular hepatocytes expressing the highest levels of LT. Magnification × 56.
LT’s C-terminus remains indispensable for SV40-induced hepatocarcinogenesis in the absence of PTEN
To determine if the failure of 17kT to induce liver tumors reflected the inability of LT’s N-terminus to breach the oncogenic threshold for hepatocyte transformation or the absence of specific transforming elements in LT’s C-terminus, we lowered the oncogenic threshold by introducing the 17kT or LT-expressing transgenes into a liver-specific PTEN-null (Δ-PTEN) background, an environment that is, by itself, permissive for hepatocarcinogenesis.28, 29 Upon obtaining mice with the requisite genotypes, cohorts of mice were placed on a 10 μg/ml dox regimen at ∼4 weeks of age to induce transgene expression and were monitored bi-weekly for the development of hepatomegaly and tumor development. Remarkably, dox-naive LT mice on a PTEN-deficient background developed hepatomegaly after 4–6 weeks (Figures 6a and b) and multi-focal HCC by ∼10 weeks of age (Figure 6b). Providing mice with dox not only increased the magnitude of LT-dependent hepatomegaly in the PTEN-deficient background after ∼2 weeks (Figure 6b), but also accelerated the development of HCC relative to PTEN-proficient mice (Figure 6c). This strong cooperation between LT and PTEN-deficiency was in complete contrast to 17kT, whereby PTEN-deficiency failed to have any effect on hepatic mass in the absence of dox (data not shown). PTEN-deficiency did, however, accelerate dox-dependent 17kT-induced hyperplasia (Figures 6d and e) and eventually promoted the development of solitary tumors in 2 of 12 mice, but only after >6 weeks of dox treatment, most likely representing tumors that, again, developed from the selective outgrowth of LT-expressing hepatocytes. Subsequent analysis showed that although PTEN-deficiency failed to impact hepatocyte apoptosis (data not shown) or induce further alterations in hepatic differentiation in either transgenic line, PTEN-deficiency enhanced 17kT- and LT-dependent hepatocyte proliferation (Supplementary Figure S7 and Supplementary Results). Moreover, this appeared to be due, in part, to the increased production of the viral oncoproteins (Supplementary Figure S8).Thus, the ability of PTEN-deficiency to cooperate with LT, but not 17kT for tumor development in the liver demonstrates that LT’s carboxy-terminus encodes oncogenic elements that are absolutely required for hepatocyte transformation, one of which most likely includes the ability to bind p53.

Loss of PTEN sensitizes livers to LT-, but not 17kT-dependent HCC. (a) Graph of liver growth, (b) gross appearance and (c) Kaplan–Meier curve of tumor-free incidence in LT-expressing livers with and without PTEN. Note that loss of PTEN (Δ-PTEN) accelerates hyperplastic growth and HCC development in LT livers even in the absence of dox. (d) Graph of liver growth and (e) gross liver appearance of 17kT-expressing livers with and without PTEN. Loss of PTEN accelerates hyperplastic growth in 17kT livers, but does not sensitize to HCC. For (a), n⩾4 mice/group; for (d), n⩾5 mice/group. Error bars +/− s.e.m. ** P<0.05, ***P<0.001.
Discussion
The unique line of transgenic mice co-expressing 17kT and st described in this report not only provided us with an opportunity to investigate the function of the naturally occurring 17kT protein in normally quiescent hepatocytes in vivo, but by comparing it with an equivalent line of mice expressing LT, we were also able to define functions and identify hepatic-specific genes and molecular signatures regulated by LT’s N-terminus in the presence or absence of LT’s C-terminus. Comparative analysis of both strains of mice demonstrated that despite the ability of 17kT and LT to commonly disrupt hepatocyte quiescence, drive hepatic hyperplasia and suppress hepatic function, only LT was capable of promoting hepatocarcinogenesis indicating that LT’s C-terminus harbors oncogenic functions that are absolutely required for hepatocyte transformation. In addition to these shared signatures, we also identified a unique 17kT-associated signature that was of particular interest due to the fact that many of the 17kT-induced genes were known transcriptional targets of p53. This was significant in that it indicated that 17kT-induced hepatic hyperplasia was not only driven by E2F-dependent cell cycle activation, but also required an override of a p53-dependent checkpoint that is normally triggered by loss of Rb.18, 19 Moreover, the fact that this signature was evident in 17kT-, but not LT-expressing livers, indicates that this signature is dominantly suppressed by LT’s C-terminus, consistent with LT-mediated abrogation of p53-dependent transcription.20, 21 While 17kT and LT both perturbed the expression of a multitude of transcriptional targets that reflected the action of LT’s N-terminus on Rb-E2F-dependent control of both universally expressed and hepatic-specific mRNAs,27, 30 evidence suggests that the 17kT-associated p53 signature is somewhat cell-type dependent as a similar p53-associated signature has been reported in mouse embryo fibroblasts expressing the N-terminal LT mutant (LT1–136), but not in intestinal enterocytes expressing LT1–136.26 Although the exact mechanism by which LT’s N-terminus overrides the p53-dependent checkpoint is not well understood, it is thought to require binding of LT’s N-terminus to Rb and/or the transcriptional co-activator p300.31, 32, 33 Thus, the 17kT-specific signature not only reflects activation of p53 in response to loss of Rb, but also likely reflects the transcriptional output of the interaction of LT’s N-terminus with Rb/p300.
Despite the fact that 17kT disrupted Rb-E2F-mediated transcriptional control, overrode the p53-dependent checkpoint and suppressed hepatic function, 17kT failed to promote tumor development in the liver, even in the absence of PTEN. This was in direct contrast to LT, which induced hepatic dysplasia, predisposed to tumorigenesis and induced a striking ‘re-programming’ signature. While a shift from an adult to a fetal gene-expression program is consistent with the idea that dysplasia and tumor predisposition are driven by altered differentiation and re-programming,34 we emphasize that this signature was derived from dysplastic livers rather than tumors, although preliminary analysis of individual tumors for several of the most highly induced LT-specific mRNAs indicates conservation between dysplastic livers and tumors in terms of altered gene expression (unpublished data). We also note that 17kT and LT both suppressed a multitude of mRNAs involved in the regulation of hepatic function and metabolism, yet only LT altered hepatic differentiation, suggesting that hepatic function and differentiation are independently and coordinately regulated by LT’s domains to simultaneously optimize proliferative capacity while reducing differentiation potential.
Although many cellular proteins bind to LT’s C-terminus,1 we propose that the LT/p53 interaction is not only the predominant driver of hepatic dysplasia and tumorigenesis in this model,35 but is also responsible for the re-programming of hepatic gene expression. For instance, LT not only suppressed the expression of a subset of p53-dependent targets and other mRNAs that were induced by 17kT, but afp and H19, two of the most highly LT-regulated transcripts, are targets of p53-mediated transcriptional repression,36, 37, 38providing additional evidence that p53-dependent transcription was compromised by LT. In addition, while p53 stabilization was clearly not required for 17kT-mediated override of the p53-dependent checkpoint, the striking correlation between stabilized p53 and high LT expression in tumor nodules provides compelling evidence that the LT/p53 interaction confers an oncogenic advantage for hepatocytes. Indeed, evidence suggests that the LT/p53 interaction provides a gain-of-function mechanism for LT-dependent transformation in certain cell types,22, 23, 39 providing an explanation as to why forced expression of WT p53does not prevent LT-induced tumorigenesis35 and why ablation of Rb and p53 is not sufficient to drive hepatocarcinogenesis in mice.40 We also propose that the re-programming of hepatocyte gene expression by LT’s C-terminus is directly related to the ability of LT to improve the efficiency of somatic re-programming,41 either by inactivating p53,42, 43 or by inducing the pluripotency factor c-Myc,44 which was markedly elevated in LT-expressing livers. However, as p300 and CBP bind to the LT/p53 complex45, 46, 47, 48, 49 and LT and the related Adenovirus E1A protein promote epigenetic re-programming via p300/CBP,50, 51 it remains to be determined to what extent the LT-associated ‘re-programming’ signature reflects the transcriptional output from various LT-containing complexes.
Finally, although our data showing that 17kT is unable to fix the transformed state in hepatocytes when co-expressed with st or in the absence of PTEN is consistent with the weak transforming activity of 17kT in vitro,4, 7the reduced transforming activity of truncated nuclear LT mutants27, 52, 53 and the absence of tumor development in Rb-deficient livers,30 it contrasts sharply with the development of HCC in transgenic mice expressing the LT mutant dl1137.54 However, as 17kT is localized to the nucleus4 and dl1137 lacks LT’s nuclear localization signal and 17kT’s C-terminal 14 amino acids, it is possible that both the proteins are functionally distinct. In speculating as to what the function of 17kT might be within the context of the SV40 virus, our data suggests that 17kT augments viral replication in the presence of LT or provides a fail-safe mechanism for DNA synthesis in its absence. Moreover, we show that unlike LT, which induces endoreduplication and promotes aneuploidy, genomic instability and dysplasia by uncoupling DNA replication from mitosis,6, 55, 56 17kT restricts DNA replication to a single round thereby maintaining coordination between DNA replication and mitosis, possibly via the induction of genes such as lmln;57 a function that may be beneficial to the virus under certain conditions. These results emphasize the need to better understand cell and context-specific responses to LT and its domains.24, 27Additional studies aimed at clarifying the role of LT’s C-terminus in cell-type dependent transformation and determining the extent to which transformation depends on formation of an LT/p53 complex or on re-programming via p300 or CBP should also help to elucidate novel pathways to transformation.
Materials and methods
Mouse strains and genotyping
ApoE-rtTAM2:TRE2-TAg transgenic mice expressing the SV40 early region under control of a liver-specific, doxycycline-inducible system were generated by co-injection of two transgenes; an ∼11 kb ApoE-rtTAM2 transgene in which the modified reverse tetracycline transactivator, rtTAs-M258 is cloned into the apoE expression cassette pLIV11,59 and a 4.5 kb TRE2-TAg transgene consisting of the 2.7 kb SV40 early region cloned into pTRE2 (Clontech, Mountain View, CA, USA) (Supplementary Figure S1). PTENlox/lox (Lesche et al.60) and Alb-Cre mice61 were obtained from The Jackson Laboratories (JAX) (Bar Harbor, ME, USA). Genotyping of mice8was conducted using primers and conditions provided in Supplementary Table S1. All mouse experiments were approved by the UT Southwestern Institutional Animal Care and Use Committee.
RT–PCR and sequencing of SV40-specific transcripts
cDNA was synthesized from DNase 1-treated total liver RNA prepared from WT and ApoE-rtTAM2:TRE2-TAg transgenic mice provided with 25 μg/ml dox in drinking water for 4 days using superscript II reverse transcriptase (Invitrogen, Grand Island, NY, USA) and random hexamer primers (Roche, Indianapolis, IN, USA). One microliter of cDNA was used as template for PCR amplification using gene-specific primers and TAq Polymerase (Qiagen, Valencia, CA, USA) (Supplementary Table S2). Confirmation of 17kT- and LT-regulated hepatic mRNAs identified by array analysis was obtained by semi-quantitative RT–PCR using primers sequences and PCR conditions described in Supplementary Table S5.
Preparation of liver proteins, liver RNA, immunoblotting and northern blotting
Preparation of liver proteins, total liver RNA, immunoblotting and northern blotting was performed as previously described in Comerford et al.8 using antibodies and reagents are listed in Supplementary Information. Northern blotting was conducted using 12 μg of total liver mRNA pooled from at least three mice on the designated regimens for each experiment.
Immunohistochemistry
IHC was performed as previously described in Clouthier et al.62 using antibodies and reagents listed in Supplementary Information. For proliferating cell nuclear antigen (PCNA), pan-cytokeratin (pan-CK) and SV40 LT IHC, microwave-antigen-retrieval in citrate buffer pH 6 (Retrievagen A, #550524, BD Biosciences) was performed for ∼15 min. Immunoreactive deposits were visualized by incubation with 3′-amino-9-ethylcarbazole (Invitrogen, 00-2007) for 2–10 min and sections were counterstained with hematoxylin QS (H-3404, Vector Laboratories, Burlingame, CA, USA).
Microarray processing and analysis
cDNA synthesis, hybridizations and all processing steps were carried out at the University of Texas Southwestern Medical Center Microarray Core Facility, Dallas, TX, USA. The 17kT- and LT-regulated genes (Supplementary Tables S3 and S4) were obtained from microarray experiments using Affymetrix MG 430 2.0 gene chips (Affymetrix, Santa Clara, CA, USA) as previously described in Schultz et al.63 Sample comparisons were performed using Genespring Version 5.1 (Silicon Genetics, Redwood City, CA, USA) and all probe sets with absent (A) calls in all samples were excluded. Lists of transcripts up- and downregulated⩾3-fold in transgenic livers relative to WT were generated for all probe sets. Changes in mRNA expression of listed genes in each table represent the average expression values for the given mRNA when multiple probe sets for the same mRNA were represented. Statistically overrepresented biological processes associated with 17kT- and LT-regulated genes were identified using Reactome Skypainter.64 Affymetrix data have been submitted to GEO; accession number GSE5242.
Accession codes
References
Ahuja D, Saenz-Robles MT, Pipas JM . SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005; 24: 7729–7745.
Cheng J, DeCaprio JA, Fluck MM, Schaffhausen BS . Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin Cancer Biol 2009; 19: 218–228.
Berk AJ, Sharp PA . Spliced early mRNAs of simian virus 40. Proc Natl Acad Sci USA 1978; 75: 1274–1278.
Zerrahn J, Knippschild U, Winkler T, Deppert W . Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J 1993; 12: 4739–4746.
DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 1988; 54: 275–283.
Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci USA 2004; 101: 947–952.
Boyapati A, Wilson M, Yu J, Rundell K . SV40 17KT antigen complements dnaj mutations in large T antigen to restore transformation of primary human fibroblasts. Virology 2003; 315: 148–158.
Comerford SA, Clouthier DE, Hinnant EA, Hammer RE . Induction of hepatocyte proliferation and death by modulation of T-Antigen expression. Oncogene 2003; 22: 2515–2530.
Kotadia S, Kao LR, Comerford SA, Jones RT, Hammer RE, Megraw TL . PP2A-dependent disruption of centrosome replication and cytoskeleton organization in Drosophila by SV40 small tumor antigen. Oncogene 2008; 27: 6334–6346.
Spangenberg HC, Thimme R, Blum HE . Serum markers of hepatocellular carcinoma. Semin Liver Dis 2006; 26: 385–390.
Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H . Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun 2006; 351: 820–824.
Kockel L, Strom A, Delacour A, Nepote V, Hagenbuchle O, Wellauer PK et al. An amylase/Cre transgene marks the whole endoderm but the primordia of liver and ventral pancreas. Genesis 2006; 44: 287–296.
Nierhoff D, Levoci L, Schulte S, Goeser T, Rogler LE, Shafritz DA . New cell surface markers for murine fetal hepatic stem cells identified through high density complementary DNA microarrays. Hepatology 2007; 46: 535–547.
Ochsner SA, Strick-Marchand H, Qiu Q, Venable S, Dean A, Wilde M et al. Transcriptional profiling of bipotential embryonic liver cells to identify liver progenitor cell surface markers. Stem Cells 2007; 25: 2476–2487.
Qiu Q, Hernandez JC, Dean AM, Rao P, Darlington G . CD24 positive cells from normal adult mouse liver are hepatocyte progenitor cells. Stem Cells Dev 2011; 20: 2177–2188.
Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM . A CD133-expressing murine liver oval cell population with bilineage potential. Stem Cells 2007; 25: 2419–2429.
Shiojiri N, Lemire JM, Fausto N . Cell lineages and oval cell progenitors in rat liver development. Cancer Res 1991; 51: 2611–2620.
Levine AJ, Hu W, Feng Z . The P53 pathway: what questions remain to be explored? Cell Death Differ 2006; 13: 1027–1036.
Pipas JM, Levine AJ . Role of T antigen interactions with p53 in tumorigenesis. Semin Cancer Biol 2001; 11: 23–30.
Bargonetti J, Reynisdottir I, Friedman PN, Prives C . Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev 1992; 6: 1886–1898.
Jiang D, Srinivasan A, Lozano G, Robbins PD . SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene 1993; 8: 2805–2812.
Hermannstadter A, Ziegler C, Kuhl M, Deppert W, Tolstonog GV . Wild-type p53 enhances efficiency of simian virus 40 large-T-antigen-induced cellular transformation. J Virol 2009; 83: 10106–10118.
Michalovitz D, Eliyahu D, Oren M . Overproduction of protein p53 contributes to simian virus 40-mediated transformation. Mol Cell Biol 1986; 6: 3531–3536.
Cantalupo PG, Saenz-Robles MT, Rathi AV, Beerman RW, Patterson WH, Whitehead RH et al. Cell-type specific regulation of gene expression by simian virus 40 T antigens. Virology 2009; 386: 183–191.
Markovics JA, Carroll PA, Robles MT, Pope H, Coopersmith CM, Pipas JM . Intestinal dysplasia induced by simian virus 40 T antigen is independent of p53. J Virol 2005; 79: 7492–7502.
Rathi AV, Saenz Robles MT, Cantalupo PG, Whitehead RH, Pipas JM . Simian virus 40 T-antigen-mediated gene regulation in enterocytes is controlled primarily by the Rb-E2F pathway. J Virol 2009; 83: 9521–9531.
Saenz Robles MT, Pipas JM . T antigen transgenic mouse models. Semin Cancer Biol 2009; 19: 229–235.
Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 2004; 113: 1774–1783.
Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci USA 2004; 101: 2082–2087.
Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ et al. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res 2005; 65: 4568–4577.
Gjoerup O, Chao H, DeCaprio JA, Roberts TM . pRB-dependent J domain-independent function of simian virus 40 large T antigen in override of p53 growth suppression. J Virol 2000; 74: 864–874.
Quartin RS, Cole CN, Pipas JM, Levine AJ . The amino-terminal functions of the simian virus 40 large T antigen are required to overcome wild-type p53-mediated growth arrest of cells. J Virol 1994; 68: 1334–1341.
Rushton JJ, Jiang D, Srinivasan A, Pipas JM, Robbins PD . Simian virus 40 T antigen can regulate p53-mediated transcription independent of binding p53. J Virol 1997; 71: 5620–5623.
Hochedlinger K, Yamada Y, Beard C, Jaenisch R . Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005; 121: 465–477.
Gillet R, Grimber G, Bennoun M, Caron de Fromentel C, Briand P, Joulin V . The consequence of p53 overexpression for liver tumor development and the response of transformed murine hepatocytes to genotoxic agents. Oncogene 2000; 19: 3498–3507.
Cui R, Nguyen TT, Taube JH, Stratton SA, Feuerman MH, Barton MC . Family members p53 and p73 act together in chromatin modification and direct repression of alpha-fetoprotein transcription. J Biol Chem 2005; 280: 39152–39160.
Dugimont T, Montpellier C, Adriaenssens E, Lottin S, Dumont L, Iotsova V et al. The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene 1998; 16: 2395–2401.
Ogden SK, Lee KC, Wernke-Dollries K, Stratton SA, Aronow B, Barton MC . p53 targets chromatin structure alteration to repress alpha-fetoprotein gene expression. J Biol Chem 2001; 276: 42057–42062.
Herzig M, Novatchkova M, Christofori G . An unexpected role for p53 in augmenting SV40 large T antigen-mediated tumorigenesis. Biol Chem 1999; 380: 203–211.
McClendon AK, Dean JL, Ertel A, Fu Z, Rivadeneira DB, Reed CA et al. RB and p53 cooperate to prevent liver tumorigenesis in response to tissue damage. Gastroenterology 2011; 141: 1439–1450.
Mali P, Ye Z, Hommond HH, Yu X, Lin J, Chen G et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells 2008; 26: 1998–2005.
Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 2011; 13: 1353–1360.
Spike BT, Wahl GM . p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer 2011; 2: 404–419.
Singhal G, Kadeppagari RK, Sankar N, Thimmapaya B . Simian virus 40 large T overcomes p300 repression of c-Myc. Virology 2008; 377: 227–232.
Avantaggiati ML, Carbone M, Graessmann A, Nakatani Y, Howard B, Levine AS . The SV40 large T antigen and adenovirus E1a oncoproteins interact with distinct isoforms of the transcriptional co-activator, p300. EMBO J 1996; 15: 2236–2248.
Borger DR, DeCaprio JA . Targeting of p300/CREB binding protein coactivators by simian virus 40 is mediated through p53. J Virol 2006; 80: 4292–4303.
Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N et al. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol 1996; 16: 3454–3464.
Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM . Binding and modulation of p53 by p300/CBP coactivators. Nature 1997; 387: 823–827.
Poulin DL, Kung AL, DeCaprio JA . p53 targets simian virus 40 large T antigen for acetylation by CBP. J Virol 2004; 78: 8245–8253.
Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK . Epigenetic reprogramming by adenovirus e1a. Science 2008; 321: 1086–1088.
Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ . Adenovirus small e1a alters global patterns of histone modification. Science 2008; 321: 1084–1085.
Pipas JM . SV40: Cell transformation and tumorigenesis. Virology 2009; 384: 294–303.
Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y et al. The amino-terminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol Cell Biol 1997; 17: 4761–4773.
Bennoun M, Grimber G, Couton D, Seye A, Molina T, Briand P et al. The amino-terminal region of SV40 large T antigen is sufficient to induce hepatic tumours in mice. Oncogene 1998; 17: 1253–1259.
Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS et al. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol 2009; 83: 117–127.
Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y et al. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev 2004; 18: 1305–1316.
McHugh B, Krause SA, Yu B, Deans AM, Heasman S, McLaughlin P et al. Invadolysin: a novel, conserved metalloprotease links mitotic structural rearrangements with cell migration. J Cell Biol 2004; 167: 673–686.
Lamartina S, Roscilli G, Rinaudo CD, Sporeno E, Silvi L, Hillen W et al. Stringent control of gene expression in vivo by using novel doxycycline-dependent trans-activators. Hum Gene Ther 2002; 13: 199–210.
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130: 1120–1133.
Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 2002; 32: 148–149.
Postic C, Magnuson MA . DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000; 26: 149–150.
Clouthier DE, Comerford SA, Hammer RE . Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest 1997; 100: 2697–2713.
Schultz N, Hamra FK, Garbers DL . A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA 2003; 100: 12201–12206.
Vastrik I, D'Eustachio P, Schmidt E, Gopinath G, Croft D, de Bono B et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol 2007; 8: R39.
Acknowledgements
We thank Mario Villarreal for animal care and gratefully acknowledge the expert services of the UTSW Molecular Pathology Core. This work was supported by funds from the Perot Family Foundation and The Excellence in Education Foundation to REH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogenesis website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Comerford, S., Schultz, N., Hinnant, E. et al. Comparative analysis of SV40 17kT and LT function in vivo demonstrates that LT’s C-terminus re-programs hepatic gene expression and is necessary for tumorigenesis in the liver. Oncogenesis 1, e28 (2012). https://doi.org/10.1038/oncsis.2012.27
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2012.27
