
Abstract
CD40, a member of the tumour necrosis factor receptor (TNFR) superfamily, has the capacity to cause extensive apoptosis in carcinoma cells, while sparing normal epithelial cells. Yet, apoptosis is only achieved by membrane-presented CD40 ligand (mCD40L), as soluble receptor agonists are but weakly pro-apoptotic. Here, for the first time we have identified the precise signalling cascade underpinning mCD40L-mediated death as involving sequential TRAF3 stabilisation, ASK1 phosphorylation, MKK4 (but not MKK7) activation and JNK/AP-1 induction, leading to a Bak- and Bax-dependent mitochondrial apoptosis pathway. TRAF3 is central in the activation of the NADPH oxidase (Nox)-2 component p40phox and the elevation of reactive oxygen species (ROS) is essential in apoptosis. Strikingly, CD40 activation resulted in down-regulation of Thioredoxin (Trx)-1 to permit ASK1 activation and apoptosis. Although soluble receptor agonist alone could not induce death, combinatorial treatment incorporating soluble CD40 agonist and pharmacological inhibition of Trx-1 was functionally equivalent to the signal triggered by mCD40L. Finally, we demonstrate using normal, ‘para-malignant’ and tumour-derived cells that progression to malignant transformation is associated with increase in oxidative stress in epithelial cells, which coincides with increased susceptibility to CD40 killing, while in normal cells CD40 signalling is cytoprotective. Our studies have revealed the molecular nature of the tumour specificity of CD40 signalling and explained the differences in pro-apoptotic potential between soluble and membrane-bound CD40 agonists. Equally importantly, by exploiting a unique epithelial culture system that allowed us to monitor alterations in the redox-state of epithelial cells at different stages of malignant transformation, our study reveals how pro-apoptotic signals can elevate ROS past a previously hypothesised ‘lethal pro-apoptotic threshold’ to induce death; an observation that is both of fundamental importance and carries implications for cancer therapy.
Similar content being viewed by others


Introduction
CD40 is a member of the tumour necrosis factor receptor (TNFR) superfamily and ligation by its cognate ligand CD40L (CD154) plays a central role in the functioning of the immune system.1 CD40 signalling primes immunocytes for humoral and cell-mediated responses and induces secretion of pro-inflammatory cytokines by epithelial cells.1, 2 A key feature of CD40-mediated signalling is the exquisite context-specificity that defines functional outcome; this is exemplified in B lymphocytes, where CD40 signalling elicits bimodal growth-regulatory effects defined by cellular differentiation and transformation stage. In resting normal B cells, CD40 signalling invokes proliferative responses, whereas activated B cells are growth-inhibited.3 Similarly, whereas CD40 signalling is mitogenic and contributes to chemotherapy resistance in low-grade B cell malignancies, in high-grade malignancies, CD40 ligation induces growth arrest and/or apoptosis.4
CD40 is expressed on a variety of cells of non-lymphoid origin, including fibroblasts, endothelial and epithelial cells and the effect of CD40 activation in such cells is equally context-specific. In addition to cytokine/chemokine secretion,5, 6 CD40 ligation may result in cell proliferation, cytostasis or apoptosis, depending on the cell type and malignant state. These observations further extend the exquisite contextual, if not paradoxical nature of CD40 signalling (discussed by Eliopoulos et al.7). The ability of CD40 to induce cytostasis or apoptosis is highly dependent on the ‘quality’ of receptor engagement. Soluble CD40 agonists (recombinant CD40L or agonistic antibody) are only cytostatic or weakly pro-apoptotic and only rendered pro-apoptotic following pharmacological intervention, for instance when combined with protein synthesis inhibitors8 or chemotherapeutic drugs.5, 9 By contrast, membrane-presented CD40L (mCD40L) is highly pro-apoptotic and induces extensive apoptosis in carcinoma cells of a variety of origins,8, 9, 10, 11, 12, 13, 14, 15, 16 irrespective of whether the ligand is presented to target carcinoma cells on the surface of third-party cells8, 13, 16 or by mCD40L-expressing naturally activated immunocytes.15 The ability of mCD40L to specifically and efficiently kill malignant cells, while sparing their normal cell counterparts, represents perhaps the most remarkable property of the CD40-mCD40L dyad.
Although CD40 lacks both death domain and intrinsic kinase activity, its activation induces a variety of signalling cascades via TRAF interacting motif (TIM) domains.17 Previous reports have associated the capacity of mCD40L to induce apoptosis over non-apoptotic soluble agonists with its ability to stabilise TRAF3 expression.11, 13 TRAF3 stabilisation allows activation of downstream mitogen activated protein kinases (MAPKs), which is dependent upon the formation of lipid rafts.18, 19 TRAF3 appears to be responsible for JNK activation following CD40L-CD40 engagement11, 13, 18 and the apoptotic potential of JNK is only reached by sustained activation during mCD40L-CD40 signalling. Thus, mCD40L may elicit a stronger or more sustained signal than soluble agonist due to its ability to invoke more efficiently a predominantly caspase 9-dependent mitochondrial pathway.11, 13 The sustained nature of JNK activation seems to be the lynchpin-determinant of pro-survival versus pro-apoptotic functions20 and JNK-mediated AP-1 induction results in apoptosis induction.21 There are two tiers of MAPK activation leading to JNK phosphorylation during cell stress, including that of MAPKKs and MAPKKKs.22 Apoptosis signalling kinase 1 (ASK1) is MAPKKK-activated in response to high levels of intracellular reactive oxygen species (ROS)23 and downstream of this, the MAPKKs MKK4 and MKK7 are often critical in apoptosis induction.21, 24 It remains unknown which of these signalling axes is involved in CD40-mediated JNK activation, or how this links to the activation of the intrinsic (mitochondrial) apoptotic pathway (discussed in Georgopoulos et al.13).
Collectively, in epithelial cells it appears that the outcome of CD40 signalling depends on both the quality of CD40 receptor engagement and the downstream threshold for apoptosis induction that is influenced by malignant transformation status.8, 16 To understand the underpinning mechanisms for these observations, we have utilised a well-characterised epithelial cell culture system that permits study of carcinoma cells, their normal epithelial counterparts, as well as isogenic ‘para-malignant’ cells, i.e. cells carrying defined genetic alterations that mimic cells undergoing malignant transformation.25, 26, 27 We have identified a ROS level-dictated, pro-apoptotic signalling cascade that provides a molecular explanation for these two fundamental properties of CD40. Using this unique model to directly compare normal and para-malignant cells to malignant cells, this study provides unifying molecular evidence for a redox state-dependent pro-apoptotic ‘lethal pro-apoptotic threshold’ that has been hypothesised to exist in epithelial cells.28, 29
Results
mCD40L, but not soluble CD40 agonist, initiates the mitochondrial apoptotic pathway in malignant but not normal epithelial cells
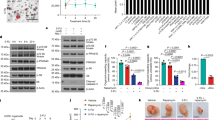
Using a co-culture system for the ligation of CD40 by mCD40L, that we had previously characterised,8, 13, 15 we confirmed that mCD40L induces apoptosis in carcinoma cells (EJ and HCT116) (Figure 1a), apoptosis involves caspase activation (Figure 1b) and cell death requires caspase activity, as death was abrogated by the pan-caspase inhibitor zVAD (Figure 1a). These assays involved co-culture of mCD40L-expressing effector cells (3T3CD40L) with epithelial cells in comparison with control co-cultures (3T3Neo effectors) and results were normalised to epithelial cell-restricted CK18 expression, as described in the Materials and Methods section, to ensure that any background from the effector cells was accounted for (Supplementary Figure 1). We additionally confirmed our findings using a DNA fragmentation assay where no background interference from the effector cells was possible during apoptosis detection (Supplementary Figure 2). Importantly, soluble agonist (cross-linked, agonistic anti-CD40 mAb G28-5 which engages the CD40 receptor similarly to soluble trimeric CD40L30) caused little detectable apoptosis in comparison to mCD40L (Figure 1c). Our results were similar when we used an independent, commercially-available agonist consisting of two adjacent CD40L trimers, MegaCD40L (not shown). In contrast to their malignant counterparts (EJ), normal human uro-epithelial (NHU) cells were completely refractory to mCD40L-mediated death; strikingly, in fact, CD40 ligation in NHU cells was reproducibly cytoprotective, as it reduced background apoptosis levels in culture (Figures 1a and b).

mCD40L initiates a caspase-dependent mitochondrial apoptotic pathway in carcinoma cells but not normal cells. (a) mCD40L induces apoptosis (expressed as fold change versus controls) in EJ and HCT116 carcinoma cells and death is caspase-mediated as demonstrated by functional blockade of apoptosis using the pan-caspase inhibitor z-VAD (50 μM). In normal (NHU) epithelial cells, mCD40L did not cause apoptosis and was cytoprotective. Bars represent mean fold change ±s.d. (n=6). Of note, a 3-4-fold change in these apoptosis detection assays corresponded to approximately ~80–85% dead cells in Annexin V-FITC/PI flow cytometry assays.8 (b) mCD40L-induced death in EJ and HCT116, but not normal NHU, cells and involves caspase-3/7 activation. Bars represent mean fold caspase activity ±s.d. (n=5). (c) Unlike mCD40L that causes extensive apoptosis in EJ and HCT116 carcinoma cells, soluble CD40 agonist (G28-5 agonistic anti-CD40 antibody) is only weakly pro-apoptotic. Bars represent mean fold change ±s.d. (n=3). (d) CD40 ligation induces Bak and Bax expression (24 h) in EJ carcinoma cells as shown by immunoblotting. By contrast, in NHU cells, a small reduction in expression of Bax and particularly Bak was observed. CK18 detection was used for the comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to confirm equal epithelial lysate loading (as explained in the Methods). Of note, although equal amounts of protein were loaded for EJ and NHU cell lysates, normal cells naturally expressed higher levels of CK18. Results are representative of three independent experiments. (e) Immunoblotting analysis of cytoplasmic (Cyto) and mitochondrial (Mito) sub-cellular fractions following mCD40L-mediated CD40 ligation (24 h) in EJ cells. Western blot confirms induction of the mitochondrial apoptotic pathway by demonstrating mCD40L-mediated cytochrome c (Cyt c) release from the mitochondria to the cytoplasm. Detection of Bcl-2 and GAPDH was used to confirm successful sub-cellular fractionation. Results are representative of at least two independent experiments.
We determined that CD40 ligation by mCD40L caused rapid induction of pro-apoptotic proteins Bak and Bax in EJ carcinoma cells as early as 12 h post-ligation (not shown) and was sustained at 24 h (Figure 1d). By contrast, no such effect was observed in normal (NHU) cells, where a small reduction in Bak (and to a lesser extent Bax) basal expression was detected following CD40 ligation with mCD40L (Figure 1d), in agreement with the lack of apoptosis and cytoprotection observed above (Figures 1a and b). Cell fractionation experiments demonstrated that mCD40L induced mitochondrial outer membrane permeabilisation (MOMP), evident by the release of mitochondrial cytochrome c into the cytoplasm. Moreover, mCD40L caused down-regulation of the anti-apoptotic mitochondrial protein Bcl-2 (Figure 1e). Unlike mCD40L, soluble CD40 agonists did not induce Bak/Bax expression or MOMP (not shown). Therefore, combined with our previous findings that CD40 ligation does not involve signalling cross-talk with any known TNF death receptors and/or their ligands,13 mCD40L-mediated apoptosis operates directly via the intrinsic mitochondrial pathway.
Activation of the JNK/AP-1 pathway and subsequent JNK/AP-1-mediated upregulation of Bak and Bax are essential in mCD40L-induced apoptosis
To determine the mechanism of Bak and Bax up-regulation as well as ascertain the functional importance of these two pro-apoptotic proteins, we examined whether the JNK/AP-1 pathway was critical, as we have previously shown that JNK phosphorylation and JNK-mediated downstream AP-1 activation are essential in mCD40L-induced death,13 while JNK and AP-1 are often associated with pro-apoptotic Bcl-2 protein expression.21 Pharmacological inhibition of the AP-1 transcription factor and, particularly, inhibition of JNK abrogated CD40 killing (Figure 2a). We also found that inhibition of JNK and AP-1 function diminished basal expression of Bak and particularly Bax protein and caused complete blockade of Bak and Bax induction following CD40 ligation (Figure 2b), thus demonstrating that induction of Bak and Bax is fully-dependent on the JNK/AP-1 pathway. To confirm that Bak and/or Bax were functionally involved in apoptosis, we prepared EJ carcinoma cells stably expressing anti-Bak or anti-Bax short hairpin RNAs (shRNAs). The shRNAs down-regulated basal protein expression and prevented mCD40L-mediated Bak and Bax induction, respectively (Figure 2c), while knockdown of both Bak and Bax resulted in blockade of CD40-mediated death (Figure 2d).

JNK/AP-1-induced Bak and Bax proteins are essential in CD40-mediated apoptosis. (a) mCD40L-mediated apoptosis in EJ carcinoma cells is dependent on the activation of the JNK kinase and the AP-1 transcription factor as indicated by the blockade and attenuation of cell death by the JNK inhibitor SP600125 (25 μM) and the AP-1 inhibitor NDGA (25 μM), respectively, in comparison to vehicle control (CON). Bars represent mean fold change ±s.d. (n=5). (b) Immunoblotting analysis demonstrates that the induction of Bak and Bax protein expression following CD40 ligation (24 h) is dependent on JNK and AP-1, as the use of both JNK inhibitor SP600125 (25 μM) and AP-1 inhibitor NDGA (25 μM) abrogates the induction of both Bak and Bax, in comparison to the untreated controls (CON). CK18 detection served as a loading control. Results are representative of three independent experiments. (c) Retrovirus transduction was used to prepare EJ cell derivatives EJ-Bak-KD and EJ-Bax-KD that stably express anti-Bak and anti-Bax shRNAs, respectively, as well as their isogenic controls (EJ-Con). Successful knockdown was confirmed by Western blotting for Bak and Bax in the appropriate cell lines following CD40 ligation (24 h), as Bak and Bax up-regulation is prevented in the EJ-Bak-KD and EJ-Bax-KD lines, respectively, in comparison to the control shRNA-expressing EJ-Con cells. CK18 detection served as a loading control. Results are representative of two independent experiments. (d) CD40-mediated apoptosis is abrogated by Bak and Bax knockdown as indicated by the blockade of caspase-3/7 activation in the EJ-Bak-KD and EJ-Bax-KD lines, in comparison to EJ-Con. Bars represent mean fold caspase activity ±s.d. (n=5).
Induction of TRAF3 and phosphorylation of ASK1 and MKK4 (but not MKK7) are essential for JNK activation and mCD40L-mediated apoptosis
Having previously demonstrated that CD40 ligation by mCD40L (but not soluble agonist) rapidly induces TRAF3 expression at the post-transcriptional level by inhibiting TRAF3 degradation and shown that both TRAF3 and JNK are essential in apoptosis,13 we investigated the pathway that underpins CD40 apoptosis in order to identify receptor proximal and more distal signalling components. As a candidate MAPKKK that might participate in signalling, we investigated the involvement of ASK1 due to its role as a pro-apoptotic factor in other systems, while downstream activation of MKK4 and MKK7 was also examined since these two MAPKKs are well established activators of JNK during the induction of apoptosis.21
Ligation of CD40 by mCD40L in carcinoma cells caused rapid induction of TRAF3 expression at 1.5 h (not shown) and particularly 3 h (Figure 3a). Stabilisation of TRAF3 coincided with phosphorylation of ASK1 (3 h) and this was followed by phosphorylation of MKK4 and JNK (6 h); however no detectable changes in MKK7 phosphorylation were observed. We confirmed that the activation of this MAPKKK-MAPKK-MAPK cascade was followed by the induction of Bak and Bax at 12 h (not shown) and 24 h (Figure 3a). No changes in expression of total ASK1, MKK4/7 or JNK were observed. By contrast, TRAF3 knockdown abrogated the activation of ASK1, subsequent phosphorylation of MKK4 and JNK, and the induction of Bak and Bax expression, confirming the critical role of TRAF3 in defining the outcome of CD40 signalling.

Induction of TRAF3 and phosphorylation of ASK-1 and MKK4 are essential in JNK-activation and CD40-mediated apoptosis. (a) Retrovirus transduction was used to establish the EJ derivative line EJ-TRAF3-KD, which stably expresses an anti-TRAF3 shRNA, and its isogenic control shRNA-expressing cell line EJ-Con. Immunoblotting experiments demonstrate that ligation of CD40 in the control cells caused induction of TRAF3 expression (3 h), but this is significantly attenuated in EJ-TRAF3-KD cells. Further Western blotting experiments show that in EJ-Con, CD40 ligation triggers the phosphorylation of ASK-1 (3 h) as well as MKK4 (6 h) and JNK (6 h). No visible changes in MKK7 phosphorylation (6 h) were observed. These changes are followed by the induction of Bak and Bax (24 h). By contrast, TRAF3 knockdown abrogates the activation of ASK-1, subsequent phosphorylation of MKK4 and JNK, as well as induction of Bak and Bax. CK18 detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to ensure equal loading. Time points are indicated on the right hand side for clarity. Results are representative of three independent experiments. Of note, in all immunoblotting experiments, expression of cytokeratin proteins (CK8 or CK18) was employed to confirm equal loading rather than total unphosphorylated protein (as explained in the Methods). (b) Retrovirus transduction was used to establish EJ cell line derivatives EJ-ASK1-KD, EJ-MKK4-KD and EJ-MKK7-KD that stably express anti-ASK-1, anti-MKK4 and anti-MKK7 shRNAs, respectively. Immunoblotting experiments in these cell lines demonstrated that CD40-mediated phosphorylation of ASK-1 (3 h) and MKK4 (6 h) is blocked by the respective shRNAs when compared to their isogenic control cells (EJ-Con – as shown in (a) above as well as in Supplementary Figure 8), while basal phospho-MMK7 expression (6 h) is reduced. CK18 detection was used to assess equal loading. Results are representative of at least two independent experiments. (c) Western blots for the detection of phosphorylated JNK following CD40 ligation (6 h) in EJ-ASK1-KD, EJ-MKK4-KD and EJ-MKK7-KD demonstrate that shRNA-mediated knockdown of ASK-1 and MKK4, but not MKK7, significantly attenuates CD40-mediated p-JNK induction observed in control cells (EJ-Con in Figure 2a above). CK18 detection was used to assess equal loading. Results are representative of two independent experiments. (d) CD40-mediated apoptosis is abrogated by TRAF3, MKK4, ASK-1 knockdown as indicated by the blockade of caspase-3/7 activation in EJ-TRAF3-KD, EJ-ASK1-KD and EJ-MKK4-KD cells, in comparison to EJ-Con. By contrast, MKK7 knockdown had no effect on apoptosis. Bars represent mean fold caspase activity ±s.d. (n=5). Results are representative of three independent experiments. (e) Immunoblotting experiments demonstrate that ligation of CD40 in HCT116 cells triggers upregulation of TRAF3 (3 h), as well as phosphorylation of MKK4 (6 h) and JNK (6 h), which is followed by the induction of Bax (12 h). CK8 detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to confirm equal loading. Results are representative of at least two independent experiments. (f) Retrovirus transduction was used to establish the HCT116 derivative lines HCT116-TRAF3-KD and HCT116-Bax-KD, which stably express anti-TRAF3 and anti-Bax shRNA, respectively, and their isogenic control shRNA-expressing cell line HCT116-Con. CD40-mediated apoptosis is attenuated by TRAF3 and Bax knockdown as indicated by the blockade of caspase-3/7 activation in HCT116-TRAF3-KD and HCT116-Bax-KD cells, in comparison to HCT116-Con. Bars represent mean fold caspase activity ±s.d. (n=5).
We then sought to ascertain the functional importance of ASK1 in this pathway and to confirm that MKK4 rather than MKK7 was responsible for subsequent JNK activation. Using kinase-specific shRNAs to knock down expression of ASK1, MKK4 and MKK7, respectively (Figure 3b), we found that loss of either ASK1 or MKK4, but not MKK7, prevented JNK phosphorylation (Figure 3c) while TRAF3 levels remained unaffected (not shown), thus suggesting that downstream of TRAF3, mCD40L-mediated signalling activates ASK1 and subsequently MKK4 to activate JNK, while phospho-MKK7 is neither induced by (Figure 3a) nor does it participate in CD40-mediated JNK activation (Figure 3c). Equally importantly, knockdown of TRAF3, ASK1 and MKK4 but not MKK7 blocked CD40-mediated apoptosis as indicated by near complete abrogation of effector caspase 3/7 activation (Figure 3d). Finally, to confirm our observations on the identity of the key components of the mCD40L signalling pathway in carcinoma cells, we investigated the role of these signalling components in an independent cell line. Similarly to bladder EJ cells, we found that CD40 ligation in colorectal adenocarcinoma HCT116 cells stabilised TRAF3 (Figure 3e), phosphorylated ASK1 but not MKK7 (not shown), induced phosphorylation of MKK4 and upregulated pro-apoptotic Bcl-2 member Bax (Figure 3e), while stable shRNA knockdowns confirmed that CD40 operated via a similar TRAF3-driven pathway that depended on the activation of the intrinsic (mitochondrial) pathway to induce cell death and caspase activation (Figure 4f).

mCD40L-induced ROS release is essential for apoptosis and is driven by TRAF3- and Nox-dependent signalling. (a) The levels of ROS in EJ and HCT116 carcinoma cells were measured by H2DCFDA labelling, which was detected spectrophotometrically and expressed as fold change relative to H2DCFDA-labelled untreated cells. Unlike cells treated with soluble agonist (10μg/ml cross-linked G28-5), mCD40L-treated cells showed rapid induction of ROS (3 h) which was blocked by the ROS scavenger/antioxidant NAC (30 mM). Bars represent mean fold increase in ROS ±s.d. (n=6). In addition to spectrophotometric measurements, ROS induction by CD40 ligation was also detected by H2DCFDA labelling and flow cytometry, and representative results for HCT116 cells from at least two experiments are shown on the overlay histogram on the right to compare controls (Con) versus mCD40L-treated cells (mCD40L). (b) mCD40L-mediated apoptosis in EJ and HCT116 carcinoma cells is blocked by both the antioxidant NAC (30 mM) and the pharmacological Nox inhibitor DPI (0.25 or 0.125 μM in EJ and HCT116, respectively). Bars represent mean fold change ±s.d. (n=6). (c) Immunoblotting experiments demonstrate that ligation of CD40 in EJ (as shown in EJ-Con cells) and HCT116 cells triggers phosphorylation of the Nox subunit p40phox. The experiments also show that CD40-mediated phosphorylation of p40phox is attenuated in TRAF3 shRNA-expressing cells (EJ-TRAF3-KD) in comparison to isogenic controls (EJ-Con). Cytokeratin detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) in EJ derivatives (CK18) and HCT116 (CK8) cells, respectively, to ensure equal loading. Results are representative of at least two independent experiments. (d) Blockade of Nox by the pharmacological inhibitor DPI (0.125 μM) completely inhibited mCD40L-mediated induction of Bak (24 h) in HCT116 cells. CK8 detection was used to assess equal loading. Results are representative of two independent experiments. (e) Western blotting shows that pharmacological inhibition of Nox by DPI (0.25 μM) blocks activation of ASK-1 (3 h) and subsequently Bax induction (24 h) by CD40 in EJ cells. CK18 detection confirmed equal loading. Results are representative of three experiments. (f) Nox blockade during CD40 ligation in HCT116 and EJ cells by treatment with DPI (as described in panels D and E, respectively), attenuated the induction of TRAF3 by mCD40L. Cytokeratin detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) in EJ (CK18) and HCT116 cells (CK8), respectively, to ensure equal loading. Results are representative of two independent experiments.
mCD40L-mediated induction of reactive oxygen species (ROS) is essential for apoptosis in carcinoma cells and is dependent on signalling mediated through TRAF3 and NADPH oxidase (Nox)
As demonstrated in elegantly performed studies by Ichijo and colleagues, the activation of ASK1 and subsequent induction of apoptosis is dependent on its release from its biological inhibitor Thioredoxin (Trx), following oxidation of Trx by reactive oxygen species (ROS).23 Despite evidence supporting a role for ROS in signalling and apoptosis triggered by the death receptor subgroup of the TNFR family,31 the role of ROS in CD40 signalling remains largely unknown particularly in epithelial cells.32 Therefore, we examined whether CD40 signalling regulates cellular ROS levels in order to activate ASK1 and provoke subsequent apoptosis.
We first compared the capacity of pro-apoptotic mCD40L and non-apoptotic CD40 agonist (cross-linked G28-5 mAb)8, 13 to induce ROS production in carcinoma (EJ and HCT116) cells. We found that mCD40L, but not soluble agonist, induced substantial ROS production (Figure 4a and Supplementary Figure 3) and this could be blocked by the ROS scavenger/antioxidant N-acetyl-L-cysteine (NAC) (Figure 4a). Of note, the G28-5 antibody is active in these cells, as evident by its ability to induce IL-8 secretion to the same extent as mCD40L.12 ROS production was essential for apoptosis, as treatment with NAC during CD40 ligation completely inhibited mCD40L-mediated death (Figure 4b). The importance of ROS was confirmed by showing that inhibition of ROS with an additional, independent antioxidant (propyl gallate) dose-dependently blocked cell death (Supplementary Figure 6). Although mitochondria are a major source of ROS, the release of ROS occurred before evidence of apoptosis became apparent, indicating that the NADPH oxidase (Nox) protein complex might be involved, as Nox represents a major source of ROS generation for secondary messenger functions.33 Blockade of Nox fully abrogated both mCD40L-mediated ROS induction (Supplementary Figure 7) and mCD40L-mediated apoptosis, as seen with NAC (Figure 4b), thus confirming that the initial ROS trigger required for apoptosis was Nox-generated. A previous report provided evidence that CD40 ligation in B cells can activate Nox and ROS production via recruitment and activation of the regulatory (activatory) Nox-2 subunit p40phox to the CD40 cytoplasmic tail.34 Interestingly, mCD40L, but not soluble CD40 agonist (not shown), induced phosphorylation of p40phox in both EJ and HCT116 carcinoma cells (Figure 4c) and in line with these observations, inhibition of Nox blocked activation of pro-apoptotic molecules such as Bak (Figure 4d). We confirmed that the p40phox/Nox pathway was the trigger for the ASK1-induced apoptotic pathway, as Nox blockade fully inhibited phosphorylation of ASK1 and pro-apoptotic Bax (Figure 4e).
We next examined the functional importance of TRAF3 in the recruitment and activation of p40phox to initiate ROS and subsequently pro-apoptotic signalling. TRAF3 knockdown attenuated mCD40L-mediated p40phox phosphorylation (Figure 4c), thus confirming the critical role of TRAF3 in p40phox activation. Therefore, in light of the observation that over-expressed TRAF3 can interact with p40phox,34 our findings provided evidence in epithelial cells that endogenously activated TRAF3 by mCD40L-triggered CD40 ligation can drive Nox activation by p40phox phosphorylation. Strikingly, we also found that Nox itself could influence the fate of TRAF3, as Nox inhibition prevented CD40-mediated TRAF3 induction (Figure 4f), thus suggesting the existence of a feedback loop between TRAF3 and Nox. Notably, unlike Nox blockade, ROS inhibition (using NAC) did not prevent TRAF3 induction (not shown) indicating that it is the direct (physical or functional) involvement of p40phox/Nox and not just ROS induction per se that is necessary for TRAF3 stabilisation.
CD40 ligation by mCD40L differentially modulates Thioredoxin-1 (Trx-1) protein levels in normal epithelial versus carcinoma cells
Our findings suggest that mCD40L-induced apoptosis requires TRAF3-induced Nox activation via p40phox phosphorylation (Figure 4) to trigger a pathway that operates via an ASK1-MKK4-JNK signalling axis (Figure 3). CD40-mediated apoptosis is ROS- and ASK1-dependent and it is well established that ASK1 activation requires ROS-mediated Trx inactivation,23 while a direct interaction between p40phox and Trx has been reported.35 We therefore examined whether CD40 ligation by mCD40L had any effect on the expression of Trx-1, the main Trx subunit regulating ASK1 function.
We determined the relative amounts of Trx-1 in carcinoma cells following CD40 ligation compared to control (untreated) cells. Interestingly, basal Trx-1 protein expression increased progressively in carcinoma cells with time in culture (Figure 5a). Strikingly, CD40 ligation by mCD40L fully (EJ cells, Figure 5a) or partially (HCT116 cells, Figure 5c) inhibited the induction of Trx-1 by actively inhibiting Trx-1 expression in carcinoma cells, and this down-regulation of Trx-1 temporally coincided with the activation of ASK1 (Figure 5b). By contrast, CD40 engagement by non-apoptotic, soluble CD40 agonists (agonistic anti-CD40 mAb or MegaCD40L) did not inhibit Trx-1 expression (not shown). Importantly, this modulation of Trx-1 expression by CD40 was cancer cell-specific, as we could only detect relatively low-level Trx-1 expression in normal (NHU) epithelial cells and receptor ligation by mCD40L had no detectable effect on Trx-1 expression (Figure 5c). Of note, the inhibition of Trx-1 expression was dependent on TRAF3 activation, as TRAF3 knockdown appeared to prevent Trx-1 inhibition (Figure 5d).

CD40 ligation by mCD40L down-regulates Trx-1 protein levels in carcinoma but not normal epithelial cells. (a) Trx-1 protein expression in EJ carcinoma cells was monitored by immunoblotting at the indicated time points following CD40 ligation. Although Trx-1 expression increased in cultures following resumption of growth after sub-culture (as seen in the controls, ‘–’), CD40 ligation by mCD40L fully blocked Trx-1 expression (as seen in ‘+’ samples). CK18 detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to ensure equal loading. Results are representative of three independent experiments. (b) Immunoblotting experiments show that CD40 ligation triggers the phosphorylation of ASK-1 at 3 h which coincides with a reduction in Trx-1 expression (the results represent co-cultures of EJ cells with mCD40L-expressing 3T3CD40L and control 3T3Neo fibroblasts). 3T3CD40L- (3T3CD40L) and 3T3Neo-only (3T3Neo) cultures showed no ASK-1 phosphorylation and EJ cells cultured alone (EJ alone) showed little (basal) ASK-1 phosphorylation. β-actin detection was used to ensure equal loading. Results are representative of two independent experiments. (c) Trx-1 protein expression was determined in normal (NHU) and malignant epithelial cells (EJ and HCT116) following treatment with mCD40L (24 h) by immunoblotting. In contrast to malignant cells where CD40 ligation induced Trx-1 reduction, NHU cells expressed low levels of Trx-1 and CD40 appeared to have no effect on Trx-1 expression. CK18 (EJ) and CK8 (HCT116) detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to demonstrate equal loading. Of note, as NHU cells naturally express higher levels of CK18 in comparison to malignant (EJ) cells, and to ensure that any differences in basal Trx-1 protein expression were not artefactual, correct loading was confirmed for urothelial cells by detection of β-actin, the levels of which were similar in both cell types. Results are representative of two independent experiments. (d) Using immunoblotting, the levels of Trx-1 protein were determined in EJ-TRAF3-KD and their isogenic controls (EJ-Con) following treatment with mCD40L (6 h). In contrast to control cells where CD40 ligation induced a reduction in Trx-1 expression, EJ-TRAF3-KD cells maintained normal Trx-1 expression. CK18 detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) to ensure equal loading. Results are representative of two independent experiments.
Trx-1 inactivation renders CD40 signalling from non-apoptotic CD40 agonist quantitatively and qualitatively equivalent to membrane-bound CD40L
The ability of mCD40L to down-regulate Trx-1 protein levels was striking and suggested that in addition to ROS induction, which would inactivate Trx-1 to free ASK1 and permit apoptosis activation, mCD40L uniquely also down-regulates Trx-1 to diminish the capacity of malignant cells to cope with oxidative stress. Moreover, in light of the lack of p40phox activation, ROS induction or Trx-1 down-regulation by soluble CD40 agonist, we hypothesised that the inability of soluble agonists to invoke cell death could be attributed to their inability to engage CD40 sufficiently to induce TRAF313 and in parallel raise the levels of ROS required to inactivate Trx-1 and permit ASK1-mediated apoptosis (Figure 4).
To address this scenario, we tested the hypothesis that pharmacological inhibition of Trx-1 using the highly-specific, well-characterised irreversible inhibitor PX-1236, 37 would synergise with soluble CD40 agonist to induce apoptosis. Although higher concentrations of PX-12 resulted in high cytotoxicity, initial pre-titration experiments allowed us to determine concentrations of PX-12 that demonstrated minimal cytotoxicity. When used alone, Trx-1 inhibitor or soluble CD40 agonists (G28-5 mAb or MegaCD40L) induced little apoptosis; however, combinations of PX-12 inhibitor and soluble agonist were synergistic in inducing extensive apoptosis in carcinoma cells (Figure 6b), comparable to that observed following CD40 ligation by mCD40L (Figure 1). By contrast, in normal (NHU) cells, neither the Trx-1 inhibitor nor the soluble agonist caused any apoptosis, whereas their combination was slightly cytoprotective (Figure 6a) and in striking similarity to mCD40L (Figure 1a). Therefore, the combinatorial treatment of PX-12 and soluble CD40 agonist induces extensive apoptosis in carcinoma, but not normal cells, and is quantitatively equivalent to mCD40L.

Combination of pharmacological Trx-1 inhibitor and soluble CD40 agonist is pro-apoptotic, tumour-specific and functionally equivalent to mCD40L. (a) Normal epithelial cells (NHU) were treated with either Trx-1 inhibitor PX-12 (4μM) or soluble CD40 agonist G28-5 mAb (10μg/mL) alone, or with combination of the two (‘PX-12+G28-5’) and apoptosis was assessed (see Methods). Neither the inhibitor nor the agonist alone induced any apoptosis and nor did their combination, which was slightly cytoprotective. Bars represent mean fold change ±s.d. (n=3). (b) Combinatorial treatment of EJ and HCT116 carcinoma cells with Trx-1 inhibitor (4μM) and soluble CD40 agonist (10μg/mL) (‘PX-12+G28-5’) synergistically mediated extensive apoptosis, whereas treatment with either the inhibitor (PX-12) or agonist (G28-5) alone caused little (EJ) or relatively low (HCT116) amounts of apoptosis. Bars represent mean fold change ±s.d. (n=5). (c) Using immunoblotting, the levels of TRAF3 protein were determined in malignant epithelial cells (EJ) treated with Trx-1 inhibitor PX-12 (2μM) or soluble CD40 agonist G28-5 mAb (10μg/mL) alone, or with combination of the two (‘PX-12+G28-5’) (6 h). Solvent alone (DMSO) treated EJ cells (Control) were used as a control for experiments involving soluble agonists, whereas controls (‘–’) versus mCD40L-treated cells (‘+’) representing 3T3Neo/EJ and 3T3CD40L co-cultures (6 h), respectively, were used to detect TRAF3 protein expression following mCD40L treatment. The results demonstrate that CD40 agonist G28-5 mAb alone caused little TRAF3 induction, however its combination with PX-12 induced TRAF3 expression as strongly as did mCD40L. (d) Apoptosis triggered by the combination of Trx-1 inhibitor and soluble CD40 agonist (as above) was inhibited by the antioxidant NAC (30 mM) as well as by knockdown of ASK-1, as shown by the lack of apoptosis in ASK-1 shRNA expressing (EJ-ASK1-KD) cells in comparison to their isogenic controls (EJ-Con). Bars represent mean fold change ±s.d. (n=6).
To determine if the combinatorial treatment triggered the same molecular programme as entrained by mCD40L, we examined the key apoptosis signalling mediators identified above. Importantly, the combination of soluble CD40 agonist (G28-5 mAb) and Trx-1 inhibitor (PX-12) triggered substantial induction of TRAF3, which was equivalent to that observed with mCD40L, whereas agonist or inhibitor alone caused little or no TRAF3 induction, respectively (Figure 6c). Moreover, similarly to mCD40L, apoptosis by combinatorial treatment was inhibited by NAC, thus confirming the importance of ROS in cell death, whilst ASK1 was essential in the induction of apoptosis as shRNA-mediated ASK1 knockdown prevented apoptosis (Figure 6d), whilst we obtained similar observations following TRAF3 knockdown (not shown). Therefore, inhibition of Trx-1 function converted the non-apoptotic CD40 signal initiated by soluble agonist into a strongly pro-apoptotic signal that caused death by an equivalent signalling mechanism to that triggered by mCD40L.
CD40-mediated regulation of ROS and apoptosis in ‘para-malignant’ and transformed versus normal epithelial cells provides evidence for a critical redox state-associated pro-apoptotic threshold
A feature of malignant cells is their ability to operate under oxidative stress,38 with the suggestion that basal ROS defines a ‘lethal threshold’ that determines whether cytotoxic insults induce apoptosis.29, 39 Formal demonstration of such a threshold has been hampered by the lack of appropriate epithelial models for the study of normal cells and their transformed counterparts.28 Based on the differences between pro- and non-apoptotic CD40 ligation, a critical signalling threshold must be reached before CD40 can induce death. As the ability of CD40 ligation to induce apoptosis is closely associated with malignant transformation,8, 13 it appears that this threshold is altered during carcinogenesis and our results implicate a critical role for ROS.
We exploited our well-characterised epithelial cell platform to study how malignant transformation affects the susceptibility of epithelial cells to pro-apoptotic signals8, 16, 27 and used CD40 signalling as a paradigm to demonstrate the existence of a redox state-dependent pro-apoptotic threshold. We included three well-characterised bladder cancer cell lines RT4, RT112 and EJ, which represent a spectrum of bladder cancer grades and stages. RT4 represents a non-malignant well-differentiated line, RT112 a moderately differentiated invasive cell line and EJ (T24 sub-line) cells represent highly-malignant carcinoma cells. These cell lines have been shown, both in vitro and in vivo, to faithfully recapitulate the grade and stage of the tumours of origin.25, 40 In support of accumulating evidence suggesting that during carcinogenesis cells gradually evolve to operate under oxidative stress, we found that basal levels of ROS in these cell lines correlated closely with the degree of anaplasty (malignancy), with the highest amount of ROS observed in the most malignant EJ cells and the lowest levels in the RT4 cell line (Figure 7a). When these cells were exposed to extreme ROS levels by treatment with H2O2, EJ was the most susceptible cell line and RT4 the least susceptible (Figure 7b). Importantly, EJ was also the most susceptible line to mCD40L-mediated apoptosis and RT4 the least susceptible (Figure 7c). As RT112 cells are naturally CD40-negative, we used a stable CD40-expressing derivative (RT112-CD40).13, 15 RT112-CD40 cells exhibited basal ROS levels equivalent to RT112 cells (not shown) and were moderately susceptible to mCD40L-mediated apoptosis (Figure 7c). Therefore, susceptibility to artificially-induced intracellular ROS elevation in tumour cells representing different malignant stages directly correlated with susceptibility to mCD40L-triggered apoptosis.

CD40-mediated modulation of ROS and apoptosis in normal, ‘para-malignant’ and transformed epithelial cells. (a) The basal levels of ROS in the panel of bladder cancer cell lines RT4 (well-differentiated, non-malignant), RT112 (moderately-differentiated, non-malignant) and EJ (undifferentiated, highly-malignant) were measured by H2DCFDA labelling. There was an apparent correlation between increased basal ROS expression and degree of anaplasty, with EJ>RT112>RT4 cells. Bars represent average relative fluorescence units (RFU) ±s.d. (n=3). (b) The RT4, RT112 and EJ cell lines were treated with the indicated concentrations of H2O2 and viability was assessed. The more anaplastic, malignant cells EJ exhibited the highest degree of susceptibility to oxidative stress in comparison to the highly differentiated, non-malignant RT4 cells that were the least susceptible. Bars represent mean % cell growth ±s.d. (n=5). (c) Ligation of CD40 by mCD40L in the cancer cell lines resulted in extensive apoptosis in the highly malignant EJ cells, whereas the non-anaplastic highly-differentiated RT4 cells showed relatively low susceptibility to CD40 ligation. RT112 cells that were engineered to express the CD40 receptor showed moderate susceptibility to CD40-mediated apoptosis. Bars represent mean % cell death ±s.d. (n=3). (d) The basal ROS in normal (NHU) cells, para-malignant NHU derivatives stably expressing hTERT (HU-hTERT) and the malignant cell line EJ were determined by H2DCFDA labelling and flow cytometry. Representative results of such analysis are shown in the overlay histogram (left), while the derived results for NHU, HU-hTERT, EJ and HCT116 from similar analyses are presented as average MFI (right). Basal ROS was higher in hTERT-expressing epithelial cells in comparison to their normal counterparts, but were lower than those observed in malignant (EJ and HCT116) cells. Bars represent mean MFI ±s.d. (n=3). (e) NHU, HU-hTERT, EJ and HCT116 cells were treated with the indicated concentrations of H2O2 and viability was assessed. The malignant cells EJ and HCT116 exhibited the highest degree of susceptibility to oxidative stress in comparison to normal NHU cells which were nearly completely refractory to the tested concentrations of H2O2. By contrast, the para-malignant HU-hTERT cells demonstrated significant susceptibility to oxidative stress, which was nevertheless lower than that of EJ and HCT116 cells. Bars represent mean % cell growth ±s.d. (n=5). (f) Ligation of CD40 by mCD40L caused substantial apoptosis in HU-hTERT cells, in comparison to their normal (NHU) counterparts that were refractory. Apoptosis in normal and para-malignant cells was compared to EJ and HCT116 cells. Bars represent mean fold change ±s.d. (n=5). (g) Immunoblotting experiments show that CD40 ligation induces Bak and to a lesser extent Bax expression (24 h) in para-malignant HU-hTERT cells in comparison to normal (NHU) cells. CK18 detection was used for comparison of controls (‘–’) versus mCD40L-treated cells (‘+’) and confirmed equal epithelial lysate loading. Results are representative of three independent experiments.
To provide insight as to how malignant transformation directly influences ROS, we examined basal ROS in normal (NHU) cells and compared it to their malignant counterparts, as well as immortal NHU derivatives that over-express the catalytic subunit of human telomerase (HU-hTERT cells). We have shown previously that despite being karyotypically normal, these cells have impaired p16 function and progressively lose the ability to undergo cytodifferentiation.26 Moreover, it has been suggested that hTERT may increase basal ROS to sensitise cells to pro-apoptotic signals during malignant transformation.41 Notably, the lowest basal ROS was detected in normal epithelial cells and the highest in carcinoma-derived cells, while hTERT-immortalised cells demonstrated moderate expression (Figure 7d), thereby supporting the notion that hTERT can raise basal ROS. Strikingly, unlike carcinoma-derived cells which were highly susceptible to oxidative stress, normal epithelial cells were completely refractory to extreme ROS related stress (Figure 7e). By contrast, HU-hTERT cells were more susceptible to treatment with H2O2 than their normal cell counterparts but could withstand higher levels of oxidative stress than carcinoma cells (Figure 7e). Importantly, when we examined susceptibility to mCD40L-mediated apoptosis, we found that HU-hTERT cells were nearly as susceptible to apoptosis as carcinoma-derived cells (Figure 7f), with mCD40L, but not soluble agonist (not shown), engaging the intrinsic apoptotic pathway, as was evidenced by the induction of Bax and particularly Bak expression (Figure 7g). Collectively, our observations using normal, ‘para-malignant’ and carcinoma-derived epithelial cells suggest that neoplastic and malignant transformation progressively raises basal ROS and forces cells to operate under oxidative stress. As a consequence, cells progressively become more susceptible to pro-apoptotic, ROS-inducing signals, such as pro-apoptotic CD40 ligation.
Discussion
Using an epithelial cell culture system encompassing carcinoma cells of different grades and stages alongside normal epithelial counterparts and isogenic ‘para-malignant’ cells, we have provided molecular explanation for two previously-described features of CD40 ligation in epithelial cells: a) mCD40L induces extensive carcinoma cell-specific apoptosis, whereas soluble agonists do not, and b) a critical pro-apoptotic signalling threshold must be reached for apoptosis to occur, which is closely linked with malignant transformation.8, 16 We found that mCD40L, but not soluble agonist, utilises ROS elevation while concurrently down-regulating anti-oxidant responses (Trx) to efficiently kill malignant cells, by operating along a TRAF3-Nox/p40phox-ASK1-MKK4-JNK signalling axis to induce the mitochondrial apoptotic pathway (Figure 8). Using the CD40 system as a biological paradigm, we have further provided evidence for the existence of a ROS-defined ‘lethal pro-apoptotic threshold’.

CD40-mediated apoptosis in carcinoma cells. Schematic diagram of the signalling pathway that underpins mCD40L-mediated, tumour cell-specific apoptosis. Unlike CD40 engagement by soluble agonist that does not adequately activate the receptor to stabilise TRAF3 and initiate signalling, ligation of CD40 by mCD40L recruits and upregulates TRAF3 and activates p40phox to induce rapid ROS elevation – which appears to be essential for TRAF3 stabilisation. The generated ROS mediates the oxidation and inactivation of Trx (Trx-1), permitting activation of ASK-1 and subsequent phosphorylation of MKK4 and JNK to activate the JNK/AP-1 cascade which is responsible for the induction of Bax and Bak expression. This triggers the intrinsic, cytochrome c-driven, caspase-9 dependent mitochondrial pathway that drives apoptosis.
The degree of receptor cross-linking is critical in engaging the CD40 apoptotic machinery adequately to induce cell death. Despite the fact that soluble CD40 agonist is biologically active, for instance, the G28-5 antibody can induce cytokine secretion, the quality of the signal is important; both soluble agonist and mCD40L can induce equivalent maximal IL-8 secretion in carcinoma cells, yet only mCD40L induces secretion of GM-CSF.12 This indicates fundamental, signal quality-related differences. This property of CD40 appears to be an important feature shared within the TNF receptor family, as ‘‘signal quality’’ (i.e. the degree of receptor activation or cross-linking) affects or determines the outcome of receptor ligation. Highly cross-linked agonistic antibodies, cross-linked soluble recombinant ligands and particularly membrane-presented ligand (achieved by co-culture of target cells with growth-arrested, ligand-expressing third-party cells) induce a greater extent of carcinoma cell death in vitro in comparison to non-cross-linked agonists31 but no mechanistic basis is available. In the context of CD40, our present findings in combination with previous observations suggest that these differences are not only based on TRAF3 stabilisation capacity but also concomitant activation of the Nox subunit p40phox.13, 34 The ability to activate TRAF3/Nox determines whether ROS induction, and subsequently apoptosis, is achieved or not. We have therefore provided molecular evidence for the importance of the quality of CD40 receptor engagement in determining the functional outcome of receptor ligation.8, 42 By understanding why mCD40L is more pro-apoptotic in comparison to non-apoptotic soluble agonists, we have consequently been able to compensate for the inability of soluble agonist to kill efficiently by combining therapy with a Trx-1 inhibitor, which rendered the soluble agonist functionally equivalent to mCD40L. This not only has important therapeutic implications, particularly as this was achieved using a clinically-tested inhibitor, but also provides evidence that engagement of the TRAF3/Nox pathway to increase ROS is essential for apoptosis.
NADPH oxidase (Nox) is an oxidoreductase that exists as a plasma membrane complex consisting of subunits p40-, p47-, p67, p20 and gp91-phox. When the subunits are modified by events such as protein phosphorylation, they transfer electrons from NADH to oxygen to form ROS.33 p40phox has been shown to play a critical co-activatory role in Nox-mediated ROS generation, with one study demonstrating a direct interaction between TRAF3 and p40phox following CD40 over-expression in mammalian B cells.34 No previous role for p40phox has been reported in epithelial cells, but based on our results, we predict a direct TRAF3/p40phox interaction occurs following TRAF3 stabilisation on the cytoplasmic tail of CD40. TRAF3 is essential for p40phox phosphorylation and subsequent activation of ASK1, so TRAF3 and the Nox system are central in apoptosis induction. As p40phox is a Trx binding protein, also known as Trx binding protein-1 (tbp-1), it may play a role in negative regulation of Trx.35, 43 Thus, it is possible that p40phox not only mediates ROS release, but also acts as a negative regulator of Trx, which may explain why we observed a progressive decrease in Trx expression in carcinoma cells treated with mCD40L. We also observed that Nox function is critical in TRAF3 stabilisation itself, which indicates a positive feedback loop-mediated amplification of the proximal stages of CD40 signalling. Therefore, the ability of mCD40L to induce death is linked with its ability to stabilise TRAF3 and generate ROS via TRAF3-Nox activation of ASK1, while possibly utilising p40phox/tbp-1 for Trx negative regulation. Our observation that both Nox and ROS inhibition block apoptosis, but only Nox blockade interferes with TRAF3 stabilisation, suggests that ROS is critical in death signalling but not in the initial TRAF3 induction and the feedback amplification.
As part of our investigation of proximal and more distal components of the apoptotic pathway triggered by CD40, we have confirmed a critical role for TRAF3-mediated activation of JNK/AP-1 in apoptosis8, 13 and shown that TRAF3 and JNK/AP-1 regulate the expression of the pro-apoptotic Bcl-2 members Bak/Bax and the induction of MOMP. JNK can be suppressed by Gluthathione S-Transferase (GSTp) until GSTp is oxidised by ROS,44 and JNK activation is often linked to pro-apoptotic Bcl-2 protein expression.45 It is possible that CD40-mediated activation of JNK/AP-1 occurs via ROS driven mechanisms, which may cause release of GSTp and subsequent Bak-Bax expression and MOMP. JNK activation also occurs following phosphorylation by MKK4 with such a response mediated by oxidative stress.46 Caught between JNK activation for apoptosis induction and MAPKKK activation for cell proliferation, it is not surprising that MKK4 has been suggested to have both tumour suppressive and oncogenic functions.47 Similarly, transient JNK expression initiates pro-survival responses whereas sustained JNK activation drives apoptosis.20, 48, 49 Therefore, both JNK and MKK4 may represent a ‘double-edged sword’ in cancer. Although MKK7 can also be critical in activation of JNK50 and JNK activation is often regulated by MKK4 and/or MKK7 following their activation by ASK1,51, 52 we could neither detect activation of MKK7, nor did MKK7 knockdown showed any influence on mCD40L killing. Instead, we have demonstrated that MKK4 regulates mCD40L-induced apoptosis via JNK activation and this is regulated by TRAF3. It is also important to emphasise that despite the wealth of publications describing the role of critical pro-apoptotic mediators, such as ASK1 (discussed below), MKK4/7 and/or JNK and their interactions in the context of apoptotic signalling, the present study is the first to investigate the expression and functional roles of such signalling mediators in a system that a) is based on endogenous protein expression and does not involve any ‘artificial’ over-expression, and b) is performed using cells of epithelial origin.
Although the importance of ASK1 in apoptosis triggered by TNFR members has been reported,31 this is the first such demonstration for CD40. More importantly, CD40 not only activates ASK1 but concurrently causes down-regulation of Trx expression; to our knowledge a first such demonstration for a TNFR member. ASK1 is a MAPKKK that is biologically inhibited by Trx in its reduced form, but Trx oxidation by ROS permits ASK1 auto-phosphorylation at Thr845.23 Active ASK1 in turn directly phosphorylates MKK4 and leads to JNK activation.53 We now provide evidence that ASK1 is a critical mediator of mCD40L-induced apoptosis via its ability to regulate activation of MKK4 and JNK. The activation of ASK1 occurring in response to mCD40L-driven oxidative stress, in combination with active down-regulation of Trx (which appears to be a unique property of CD40), explains why soluble CD40 agonists, which have a low ROS-induction capacity, do not activate ASK1 and cannot kill carcinoma cells efficiently. Hence, when soluble agonist is combined with an inhibitor of Trx-1 and negates the requirement to raise ROS adequately to permit ASK1 activation, the non-apoptotic agonist becomes significantly pro-apoptotic, which is further supported by our finding that the combination of CD40 agonist and Trx-1 inhibitor stabilises TRAF3 as efficiently as does mCD40L. Moreover, the latter finding also suggests the possibility that when the quality of the CD40 signal is appropriate, a combination of adequate ROS elevation as well as direct involvement of mediators such as Nox/p40phox is essential; it is tempting to speculate that this involvement could be dependent on the formation of a large multi-protein complex where specific interactions (e.g. between TRAF3 and Nox) are essential. Importantly, we have provided evidence that like mCD40L-induced apoptosis, the cell death response triggered by the combination of soluble CD40 agonist and Trx inhibitor is also TRAF3-ROS-ASK1 driven, ROS-dependent and remains tumour cell-specific and, like mCD40L, this combination is cytoprotective in normal epithelial cells.
Carcinoma cells operate under conditions of oxidative stress, often as a consequence of malignant transformation.38 The potential role of basal cellular ROS in determining whether cytotoxic agents can induce apoptosis in malignant cells by ‘pushing’ them past a critical ‘lethal threshold’ has long been suggested.29, 39 There is also evidence that during the process of malignant transformation, genetic changes that drive carcinogenesis (such as the activation of oncogenes) can increase basal oxidative stress.29, 54 Overcoming the influence of ROS can determine the ability to metastasise or not55 and as a result, malignant cells are (a) obliged to upregulate antioxidant defence systems controlling redox balance,56, 57 which explains the increased expression of endogenous antioxidants such as Trx in cancer,36 and (b) progressively more vulnerable to ROS-associated insults that further augment ROS generation and/or weaken antioxidant defences in cells.38, 39 Therefore, it is clear that with such ‘cell proliferation at a cost’, tumour cells may operate in a ‘primed’ state58 that can more readily ‘push’ them past such a lethal threshold. However, it is widely accepted that lack of appropriate, representative epithelial models for the study of normal cells and their transformed epithelial counterparts has been a difficult obstacle to overcome in order to test these hypotheses.28
Our data demonstrate that the basal, steady-state intracellular ROS levels in vitro positively correlate with the tumour stage/grade and the degree of malignant transformation of cells and that an increased ROS level was associated with a more malignant phenotype. Elevated basal ROS production in more malignant cell lines rendered them more susceptible to extreme oxidative stress. By contrast, normal cells showed the lowest detectable levels of basal ROS and were highly refractory to oxidative stress. Equally, susceptibility to CD40 coincided with basal ROS levels in malignant cells and their vulnerability to extreme ROS-mediated stress. This implied a direct correlation between basal ROS and susceptibility to both CD40 and oxidative stress, suggesting that as tumour cells maintain high ROS levels to support proliferation, CD40 ligation ‘pushes’ cells past a critical ROS-related pro-apoptotic threshold to induce death. By contrast, the cytoprotective ability of CD40 ligation in normal cells was striking and in accordance with previous observations by Young and colleagues who reported that constitutive CD40 activation has the potential to transform cells (albeit fibroblasts) in vitro,59 as well as in agreement with the suggestion that in a normal cell setting ROS may induce proliferation.38 Moreover, unlike malignant cells that operate under high oxidative stress and rapidly induce Trx expression, we have now established that normal cells not only operate at lower basal ROS but also show little Trx expression. Our observation that basal Trx-1 protein expression progressively increased during culture of carcinoma cells is in support of the reported over-expression of Trx-1 in tumour cells.36
Despite reported contrasting roles for hTERT in the regulation of apoptosis and ROS induction,41 HU-hTERT cells displayed intermediate basal ROS levels and susceptibility to apoptosis by CD40 and extreme ROS stress that was substantial, albeit not equal to that of malignant cells. Of note, we have previously shown that CD40 ligation can induce a small level of apoptosis in p16-deficient (HU-CDK4mut) NHU cells.16 We found that HU-hTERT cells expressed higher levels of basal ROS and were more susceptible to CD40-mediated apoptosis than HU-CDK4mut cells (unpublished observations). Our findings on HU-hTERT cells mirror our observations on HPV E6-immortalised (HU-E6) urothelial cells. Both HU-E68 and HU-hTERT cells are highly-susceptible to CD40 death supported by the induction of pro-apoptotic Bcl-2 proteins. Interestingly, HPV E6 is a multifunctional oncoprotein that both inactivates p1660 and triggers hTERT activation.61 Our findings with p16 functional-knockout cells previously16 and hTERT over-expressers here, may provide an explanation for the observations on HPV E6 and its ability to render urothelial cells susceptible to CD40 killing. Certainly our previous observation that HU-hTERT progressively lost p16 expression26 implies that the combinatorial loss of p16 function and other hTERT-specific effects may be the two main requirements that confer CD40 susceptibility. Therefore, our findings have provided evidence for the importance of malignant transformation in rendering cells susceptible to ROS-dependent apoptosis.
In summary, our investigations have allowed us to understand the precise molecular signature of the CD40 pathway and, using it as a paradigm, explained how cellular redox state determines survival versus death decisions. Using a unique epithelial model, this study has for the first time provided evidence for a redox state, ROS-dependent ‘lethal pro-apoptotic threshold’ that has been hypothesised to exist in epithelial cells28, 38, 58 and has been suggested as a promising therapeutic target.29, 39 The evidence for this ‘double-edged sword’ nature of ROS in determining epithelial cell fate explains how oxidative stress-operating malignant cells can be more susceptible to apoptosis, which is biologically fundamental and opens new doors to cancer-selective therapies.
Materials and methods
Cell culture
Human bladder carcinoma-derived EJ cells and colorectal carcinoma-derived HCT116 cells were cultured in a 1:1 (v/v) mixture of DMEM and RPMI 1640 containing 5% FBS (Sigma, Dorset, UK), referred to as DR 5% medium. NHU cells and HU-hTERT cells were established and cultured in complete KSFM (Fisher Scientific, Loughborough, UK) as described.8, 26, 62 3T3neo and 3T3CD40L fibroblasts13, 15, 16 were cultured in DMEM supplemented with 10% FBS and containing 0.5 mg/ml G418, with omission of antibiotic in co-culture experiments (below).
Cloning of shRNAs constructs and expression in epithelial cells by retroviral transduction
For the preparation of short hairpin RNA (shRNA) for target proteins TRAF3, MKK4, MKK7, ASK1, Bax and Bak, the pSIREN-RetroQ retroviral vector system was used according to the manufacturer’s instructions (Clontech, Takara Bio Europe, Saint-Germain-en-Laye, France). 2-3 independent shRNAs were designed for each target and the constructs that showed optimal inhibition of apoptosis (Supplementary Figure 4) contained the following sequences: 5′- GAGTCAGGTTCCGATGATC-3′ (TRAF3); TAAGCTACTTGAACACAG (MKK7); CATGGAGCTGCAGAGGATG (Bax); TCCCAATCCTACAGGAGTT (MKK4); CCGACGCTATGACTCAGAG (Bak); CCCTGCATTTTGGGAAACT (ASK1). Oligonucleotides (Eurofins, Eurofins MWG Operon, London, UK) were designed to incorporate the TTCAAGAGA hairpin loop sequence, BamHI and EcoRI overhangs for cloning into pSIREN-RetroQ as well as an internal MluI restriction site to aid in selection of positive clones, as recommended by the manufacturer. Oligonucleotides were annealed and cloned to derive plasmids pSIR-TRAF3, pSIR-MKK4, pSIR-MKK7, pSIR-ASK1, pSIR-Bax and pSIR-Bak. A negative (scrambled) control (pSIR-Con) was used as supplied by Clontech (Takara Bio Europe).
PT67 fibroblasts were transfected with each plasmid using Effectene (Qiagen, Crawley, UK). Stable retrovirus-producing lines PT67-TRAF3, PT67-MKK4, PT67-MKK7, PT67-ASK1, PT67-Bax and PT67-Bak were established by selection in culture medium containing 2 μg/ml puromycin (Invivogen, Toulouse, France). Retroviral transductions were performed as described previously.12, 13 EJ cells were transduced with retrovirus collected as conditioned medium from the appropriate PT67 cell lines, followed by selection in 0.25 μg/ml puromycin resulting in the derivation of EJ-TRAF3-KD, EJ-MKK4-KD, EJ-MKK7-KD, EJ-ASK1-KD, EJ-Bax-KD and EJ-Bak-KD as well as isogenic EJ-Con (scrambled control shRNA-expressing) cells.
CD40 ligation by soluble receptor agonist and mCD40L
Normal and malignant epithelial cells were treated with the agonistic anti-CD40 mAb G28-5 at 10 μg/ml (purified from the HB-9110 hybridoma line, purchased from the ATCC) cross-linked with affinity-purified human serum protein-adsorbed goat anti-mouse IgG at 5 μg/mL as described.8 For some experiments the MegaCD40L preparation (Enzo, Exeter, UK) was also used. To activate CD40 by membrane CD40L (mCD40L), 3T3neo (control) and mCD40L-expressing 3T3CD40L fibroblasts (effector cells) were growth-arrested with Mitomycin C (Sigma) and co-cultures with epithelial (target) cells were performed as described.12, 13 3T3 cells were not growth-arrested in experiments involving ROS detection (below). 3T3 cells were seeded at 104 or 105 cells/well in 96- or 24-well plates, respectively, and epithelial cells were subsequently seeded onto 3T3 cells at a ratio of 0.8:1 for EJ cells and 1:1 for NHU and HCT116 cells. Following pre-titration experiments involving effector:target ratios 1:0.8, 1:1 and 1:1.2, optimal effector:target ratios were determined based on either apoptosis and caspase activation detection assays (below) for malignant cells and cytokine (IL-6) release measurements for NHU cells.12
Functional blocking studies with pharmacological inhibitors
The AP-1 inhibitor (NDGA), NADPH oxidase inhibitor (DPI), Thioredoxin inhibitor (PX-12), and the antioxidants N-acetyl L-cysteine (NAC) and propyl gallate were obtained from Sigma. The JNK inhibitor (SP600125) was obtained from Enzo. NAC was initially reconstituted in DR 5% culture medium at 30 mM, its pH was adjusted to 7.0 using 1M NaOH solution and was filter-sterilised before use. Other inhibitors were reconstituted in DMSO (Sigma) or ethanol (propyl gallate) and appropriate solvent-only controls were included in all experiments. For some experiments, cells were treated with hydrogen peroxide (H2O2) (Sigma) diluted in culture medium at the indicated concentrations for 48 h. The effects of H2O2 on cell growth were quantified by measuring cell biomass using the CellTiter 96 AQueous One cell proliferation assay (Promega) and results were expressed as % cell growth of each condition in comparison to untreated controls, as detailed elsewhere.63
Assessment of apoptosis and effector caspase activation
Previously published guidelines regarding the use and interpretation of assays for monitoring cell death64 have recommended that a minimum of two assays are utilised for the detection of cell apoptosis. Apoptosis was detected using CytoTox-Glo assay (Promega, Southampton, UK) and caspase-3/7 activation using the SensoLyte Homogeneous AFC Caspase-3/7 assay (Anaspec, Cambridge Bioscience, Cambridge, UK). Epithelial cells were treated with soluble CD40 agonist or mCD40L by co-culture with effector 3T3 cells in 96-well plates before apoptosis assays were performed by luminescence (Cytotox-Glo, Promega) and fluorescence (SensoLyte, Anaspec) measurements were taken on a FLUOstar OPTIMA plate reader and data analysed using MARS software (BMG Labtech, Bucks, UK). To account for background attributable to the effector cells in co-cultures, 3T3 cells were cultured alone and their relative luminescence or fluorescence units subtracted from the representative co-culture in a pair wise fashion. Results were presented as fold change or fold caspase activity, respectively, of 3T3CD40L co-cultures relative to 3T3neo co-culture controls. All assays were performed at 48 h unless otherwise stated.
Detection of ROS
6-carboxy-2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) (Fisher Scientific) was used to detect reactive oxygen species (ROS). Epithelial cells were treated with soluble CD40 agonist or mCD40L as described above. Following CD40 ligation for 3 h, cultures of agonist treated cells or co-cultures were washed with PBS and were then treated with 1 μM of H2DCFDA in pre warmed (37 °C) PBS for 30 min at 37 °C in 5% CO2. Fluorescence was then measured by either flow cytometry on a Guava EasyCyte (Millipore, Watford, UK) or by spectrophotometry on a FLUOstar OPTIMA plate reader and expressed as median fluorescence intensity (MFI) or fold change (following background subtraction) with respect to controls, respectively.
Immunoblotting (Western blot) analysis
Whole-cell lysates were prepared (Bugajska et al.,8) separated by 4-12% SDS-PAGE, followed by electroblotting using Immobilon-FL polyvinylidine difluoride (PVDF) membrane (FDR-523-020Q) (Fisher Scientific) and probed with the following antibodies: TRAF3 (sc-949/C20), cytochrome c (sc-7159) (Santa Cruz, supplied by Insight Bio, Wembley, UK), phospho-MKK4 (Ser457) (#4514 (C36C11)), phospho-MKK7 (Ser271/Thr275) (#4171), phospho-ASK1 (Thr845) (#3765), phospho-JNK (Thr183/Tyr185) (255 (G9)), Thioredoxin-1 (#2285S), phospho-p40phox (Thr154) (#4311) (Cell Signalling Technologies, supplied by New England Biolabs, Herts, UK), Bak (AF816), Bax (2282-MC-100 (YTH-2D2)), and Bcl-2 (2291-MC-100 (YTH-8C8) (R&D Systems, Abingdon, UK). It should be noted that as lysates were prepared from 3T3neo/3T3CD40L and epithelial cell co-cultures, sample loading was corrected and adjusted for epithelial cells. This was carried out according to reactivity with human-specific anti-CK18 (Sigma Scientific) or anti-CK8 (Fisher) antibodies (and not using antibodies detecting non-phosphorylated intracellular signalling mediators), as explained elsewhere.13 Antibody binding was detected by incubation with goat anti-rabbit immunoglobulin conjugated to IRDye® 800 (Tebu-bio, Cambs, UK) or goat anti-mouse immunoglobulin conjugated to Alexa Fluor 680 (Invitrogen, Paisley, UK). Immunolabelling was visualised using an Odyssey infra-red imaging system (LiCor, Cambs, UK) and densitometry performed for representative blots (Supplementary Figure 5) using the manufacturer’s (LiCor) software.
Sub-cellular fractionation
Mitochondrial and cytoplasmic fractions were isolated from co-cultures performed in 10 cm2 dishes (3 × 106 3T3 cells and 2.7 × 106 epithelial cells) for 24 h using the mitochondrial isolation kit (Millipore #MT1000) as instructed by the manufacturer. Mitochondrial pellets were lysed and processed as for the preparation of whole cell lysates. Successful mitochondrial and cytoplasmic fractionation was confirmed by detection of Bcl-2 and GAPDH, respectively. All primary antibodies were supplied with the kit (Millipore) and immunoblotting using appropriate secondary antibodies was performed as detailed above.
Statistics
Mean values and standard deviation (s.d.) were used as descriptive statistics. Two-tailed, paired or non-paired Student’s t-tests, and where appropriate ANOVA (with post tests), were used for evaluation of statistical significance. For graphical purposes *P<0.05, **P<0.01 and ***P<0.001.
References
Grewal IS, Flavell RA . CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol 1998; 16: 111–135.
Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, Noelle RJ . Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 2009; 229: 152–172.
Johnson P, Watt S, Betts D, Davies D, Jordan S, Norton A et al. Isolated follicular lymphoma cells are resistant to apoptosis and can be grown in vitro in the CD40/stromal cell system. Blood 1993; 82: 1848–1857.
Schattner E, Friedman SM . Fas expression and apoptosis in human B cells. Immunol Res 1996; 15: 246–257.
Eliopoulos AG, Dawson CW, Mosialos G, Floettmann JE, Rowe M, Armitage RJ et al. CD40-induced growth inhibition in epithelial cells is mimicked by Epstein-Barr Virus-encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene 1996; 13: 2243–2254.
Altenburg A, Baldus SE, Smola H, Pfister H, Hess S . CD40 ligand-CD40 interaction induces chemokines in cervical carcinoma cells in synergism with IFN-gamma. J Immunol. 1999; 162: 4140–4147.
Eliopoulos AG, Young LS . The role of the CD40 pathway in the pathogenesis and treatment of cancer. Curr Opin Pharmacol 2004; 4: 360–367.
Bugajska U, Georgopoulos NT, Southgate J, Johnson PWM, Graber P, Gordon J et al. The effects of malignant transformation on susceptibility of human urothelial cells to CD40-mediated apoptosis. J Natl Cancer Instit. 2002; 94: 1381–1395.
Vardouli L, Lindqvist C, Vlahou K, Loskog AS, Eliopoulos AG . Adenovirus delivery of human CD40 ligand gene confers direct therapeutic effects on carcinomas. Cancer Gene Ther 2009; 16: 848–860.
Elmetwali T, Searle PF, McNeish I, Young LS, Palmer DH . CD40 ligand induced cytotoxicity in carcinoma cells is enhanced by inhibition of metalloproteinase cleavage and delivery via a conditionally-replicating adenovirus. Mol Cancer 2010; 9: 52.
Elmetwali T, Young LS, Palmer DH . CD40 ligand-induced carcinoma cell death: a balance between activation of TNFR-associated factor (TRAF) 3-dependent death signals and suppression of TRAF6-dependent survival signals. J Immunol 2010; 184: 1111–1120.
Georgopoulos NT, Merrick A, Scott N, Selby PJ, Melcher A, Trejdosiewicz LK . CD40-mediated death and cytokine secretion in colorectal cancer: a potential target for inflammatory tumour cell killing. Int J Cancer 2007; 121: 1373–1381.
Georgopoulos NT, Steele LP, Thomson MJ, Selby PJ, Southgate J, Trejdosiewicz LK . A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ 2006; 13: 1789–1801.
Hess S, Engelmann H . A novel function of CD40: induction of cell death in transformed cells. J Exp Med 1996; 183: 159–167.
Hill KS, Errington F, Steele LP, Merrick A, Morgan R, Selby PJ et al. OK432-activated human dendritic cells kill tumor cells via CD40/CD40 ligand interactions. J Immunol 2008; 181: 3108–3115.
Shaw NJ, Georgopoulos NT, Southgate J, Trejdosiewicz LK . Effects of loss of p53 and p16 function on life span and survival of human urothelial cells. Int J Cancer 2005; 116: 634–639.
Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ . TRAF proteins in CD40 signaling.Adv Exp Med Biol 2007; 597: 131–151.
Dadgostar H, Cheng G . Membrane localization of TRAF 3 enables JNK activation. J Biol Chem 2000; 275: 2539–2544.
Xie P . TRAF molecules in cell signaling and in human diseases. J Mol Signal 2013; 8: 7.
Kamata H, Honda S-i, Maeda S, Chang L, Hirata H, Karin M . Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005; 120: 649–661.
Dhanasekaran DN, Reddy EP . JNK signaling in apoptosis. Oncogene 2008; 27: 6245–6251.
Kyriakis JM, Avruch J . Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 2012; 92: 689–737.
Soga M, Matsuzawa A, Ichijo H . Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int J Cell Biol 2012; 2012: 439587.
Davis RJ . Signal transduction by the JNK group of MAP kinases. Cell 2000; 103: 239–252.
Southgate J, Masters JR, Trejdosiewicz LK . Culture of Human Urothelium. In: 2012; Freshney RIaF MG (eds), Cult Epithel Cells. J Wiley and Sons, Inc.: New York, NY, USA, 2002, pp 381–400.
Georgopoulos NT, Kirkwood LA, Varley CL, MacLaine NJ, Aziz N, Southgate J . Immortalisation of normal human urothelial cells compromises differentiation capacity. Eur Urol 2011; 60: 141–149.
Crallan RA, Georgopoulos NT, Southgate J . Experimental models of human bladder carcinogenesis. Carcinogenesis 2006; 27: 374–381.
Schumacker PT . Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer cell 2006; 10: 175–176.
Trachootham D, Alexandre J, Huang P . Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8: 579–591.
Pound JD, Challa A, Holder MJ, Armitage RJ, Dower SK, Fanslow WC et al. Minimal cross-linking and epitope requirements for CD40-dependent suppression of apoptosis contrast with those for promotion of the cell cycle and homotypic adhesions in human B cells. Int Immunol 1999; 11: 11–20.
Albarbar B, Dunnill C, Georgopoulos NT . Regulation of cell fate by lymphotoxin (LT) receptor signalling: Functional differences and similarities of the LT system to other TNF superfamily (TNFSF) members. Cytokine Growth Factor Rev 2015; 26: 659–671.
Bhogal RH, Weston CJ, Curbishley SM, Adams DH, Afford SC . Activation of CD40 with platelet derived CD154 promotes reactive oxygen species dependent death of human hepatocytes during hypoxia and reoxygenation. PloS one 2012; 7: e30867.
Jiang F, Zhang Y, Dusting GJ . NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011; 63: 218–242.
Ha YJ, Lee JR . Role of TNF receptor-associated factor 3 in the CD40 signaling by production of reactive oxygen species through association with p40phox, a cytosolic subunit of nicotinamide adenine dinucleotide phosphate oxidase. J Immunol 2004; 172: 231–239.
Nishiyama A, Ohno T, Iwata S, Matsui M, Hirota K, Masutani H et al. Demonstration of the interaction of thioredoxin with p40phox, a phagocyte oxidase component, using a yeast two-hybrid system. Immunol Lett 1999; 68: 155–159.
Baker AF, Dragovich T, Tate WR, Ramanathan RK, Roe D, Hsu C-H et al. The antitumor thioredoxin-1 inhibitor PX-12 (1-methylpropyl 2-imidazolyl disulfide) decreases thioredoxin-1 and VEGF levels in cancer patient plasma. J Lab Clin Med 2006; 147: 83–90.
Ramanathan RK, Stephenson JJ, Weiss GJ, Pestano LA, Lowe A, Hiscox A et al. A phase I trial of PX-12, a small-molecule inhibitor of thioredoxin-1, administered as a 72-hour infusion every 21 days in patients with advanced cancers refractory to standard therapy. Invest New Drugs. 2012; 30: 1591–1596.
Gupta SC, Hevia D, Patchva S, Park B, Koh W, Aggarwal BB . Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal 2012; 16: 1295–1322.
Wang H-CR, Choudhary S . Reactive oxygen species-mediated therapeutic control of bladder cancer. Nat Rev Urol 2011; 8: 608–616.
Booth C, Harnden P, Trejdosiewicz LK, Scriven S, Selby PJ, Southgate J . Stromal and vascular invasion in an human in vitro bladder cancer model. Lab Invest 1997; 76: 843–857.
Saretzki G . Telomerase, mitochondria and oxidative stress. Exp Gerontol 2009; 44: 485–492.
Stewart R, Wei W, Challa A, Armitage RJ, Arrand JR, Rowe M et al. CD154 Tone Sets the Signaling Pathways and Transcriptome Generated in Model CD40-Pluricompetent L3055 Burkitt's Lymphoma Cells. J Immunol. 2007; 179: 2705–2712.
Yodoi J, Masutani H, Nakamura H . Redox regulation by the human thioredoxin system. Biofactors 2001; 15: 107–111.
Simic T, Savic-Radojevic A, Pljesa-Ercegovac M, Matic M, Mimic-Oka J . Glutathione S-transferases in kidney and urinary bladder tumors. Nat Rev Urol 2009; 6: 281–289.
Jin HO, Park IC, An S, Lee HC, Woo SH, Hong YJ et al. Up-regulation of Bak and Bim via JNK downstream pathway in the response to nitric oxide in human glioblastoma cells. J Cell Physiol 2006; 206: 477–486.
Kim SD, Moon CK, Eun S-Y, Ryu PD, Jo SA . Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem Biophys Res Commun 2005; 328: 326–334.
Whitmarsh AJ, Davis RJ . Role of mitogen-activated protein kinase kinase 4 in cancer. Oncogene 2007; 26: 3172–3184.
Jing LIU, Anning L . Role of JNK activation in apoptosis: a double-edged sword. Cell Res 2005; 15: 36–42.
Lei K, Nimnual A, Zong W-X, Kennedy NJ, Flavell RA, Thompson CB et al. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol Cell Biol 2002; 22: 4929–4942.
Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ . MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev 2001; 15: 1419–1426.
Cargnello M, Roux PP . Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 2011; 75: 50–83.
Wagner EF . Nebreda ÃnR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer. 2009; 9: 537–549.
Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–94.
Benhar M, Dalyot I, Engelberg D, Levitzki A . Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol 2001; 21: 6913–6926.
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015; 527: 186–191.
Young TW, Mei FC, Yang G, Thompson-Lanza JA, Liu J, Cheng X . Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res 2004; 64: 4577–4584.
Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015; 27: 211–222.
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011; 334: 1129–1133.
Baxendale AJ, Dawson CW, Stewart SE, Mudaliar V, Reynolds G, Gordon J et al. Constitutive activation of the CD40 pathway promotes cell transformation and neoplastic growth. Oncogene 2005; 24: 7913–7923.
Reznikoff CA, Yeager TR, Belair CD, Savelieva E, Puthenveettil JA, Stadler WM . Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996; 56: 2886–2890.
Duffy CL, Phillips SL, Klingelhutz AJ . Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 2003; 314: 196–205.
Southgate J, Hutton K, Thomas D, Trejdosiewicz LK . Normal human urothelial cells in vitro: proliferation and induction of stratification. Lab Invest 1994; 71: 583–594.
Al-Tameemi W, Dunnill C, Hussain O, Komen MM, van den Hurk CJ, Collett A et al. Use of in vitro human keratinocyte models to study the effect of cooling on chemotherapy drug-induced cytotoxicity. Toxicol in vitro. 2014; 28: 1366–1376.
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
Acknowledgements
The authors are grateful to all clinical colleagues for the supply of normal urothelial tissue, and would like to thank Prof Alan Melcher and Dr Liz Illett (Targeted & Biological Therapies Group, Leeds Institute of Cancer and Pathology, University of Leeds, UK) for the provision of the G28-5 antibody and fruitful discussions. JS holds a research chair supported by York Against Cancer.
Author contributions
CJD designed and performed experiments, analysed data and wrote the manuscript. KI and AM performed experiments and analysed data. JS helped conceptualise the study, designed experiments and wrote the manuscript. NTG conceptualised the study, designed experimental work, analysed data and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Dunnill, C., Ibraheem, K., Mohamed, A. et al. A redox state-dictated signalling pathway deciphers the malignant cell specificity of CD40-mediated apoptosis. Oncogene 36, 2515–2528 (2017). https://doi.org/10.1038/onc.2016.401
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.401
This article is cited by
-
Novel induction of CD40 expression by tumor cells with RAS/RAF/PI3K pathway inhibition augments response to checkpoint blockade
Molecular Cancer (2021)
-
CD40 induces renal cell carcinoma-specific differential regulation of TRAF proteins, ASK1 activation and JNK/p38-mediated, ROS-dependent mitochondrial apoptosis
Cell Death Discovery (2019)
-
FABP7 Protects Astrocytes Against ROS Toxicity via Lipid Droplet Formation
Molecular Neurobiology (2019)
-
Biomimetic artificial organelles with in vitro and in vivo activity triggered by reduction in microenvironment
Nature Communications (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}