
Abstract
Epithelial–mesenchymal transition (EMT) is a critical event in metastasis of colorectal cancer (CRC). Rho/ROCKs signaling has a pivotal role in orchestrating actin cytoskeleton, leading to EMT and cancer invasion. However, the underlying mechanisms for ROCKs activation are not fully understood. Here, we identified FOXM1D, a novel isoform of Forkhead box M1 (FOXM1) that has a pivotal role in ROCKs activation by directly interacting with coiled-coil region of ROCK2. FOXM1D overexpression significantly polymerizes actin assembly and impairs E-cadherin expression, resulting in EMT and metastasis in xenograft mouse model and knockdown of FOXM1D has the opposite effect. Moreover, a high FOXM1D level correlates closely with clinical CRC metastasis. FOXM1D-induced ROCKs activation could be abrogated by the ROCKs inhibitors Y-27632 and fasudil. These observations indicate that the FOXM1D–ROCK2 interaction is crucial for Rho/ROCKs signaling and provide novel insight into actin cytoskeleton regulation and therapeutic potential for CRC metastasis.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is one of the most common malignancies. It is the third leading cause of cancer-related deaths in the United States.1 The outcome of metastatic CRC patients is disappointing and the 5-year survival rate is <12%.2 However, the metastatic mechanism of CRC is still inadequate. It is known that epithelial–mesenchymal transition (EMT) is one of the important cellular phenomena that facilitates metastasis.3 During the EMT process, cancer cells undergo marked morphological changes via a process that is regulated by Rho family GTPases.4 When bound to GTP, RhoA/C activates the serine/threonine kinases ROCK (Rho-associated kinase) 1/2. Subsequently, ROCKs activation can orchestrate the architectural arrangement of actin cytoskeleton and/or microtubule network, resulting in alterations in cell adhesion, motility and invasion, and consequently leading to EMT and metastasis.5, 6 Conversely, Rho/ROCKs inactivation may trigger the mesenchymal–epithelial transition via cytoskeleton depolymerization.7 This inactivation has also been shown to reduce the metastasis and growth of different types of cancers in mice.8 Thus, Rho/ROCKs signaling has an essential role in the invasion of tumor cells by controlling their morphological changes and metastatic behavior.9 This highlights the importance of ROCKs activity modulation for cancer treatment. However, it still remains elusive how exactly RhoA/C activates ROCKs.
In our attempt to elucidate this mechanism, we first discovered FOXM1D as a possible regulator of the RhoA/C-ROCKs signaling pathway. Forkhead box M1 (FOXM1) belongs to a large family of conserved transcriptional regulators that are defined by a common DNA-binding domain termed the forkhead box.10 To date, three primary isoforms of FOXM1 have been identified based on alternative splicing in humans, that is, FOXM1A, FOXM1B and FOXM1C (Supplementary Figure S1a). Both FOXM1B and FOXM1C can act as transcriptional activators, whereas FOXM1A appears to function as a transcriptional repressor.11 In addition, a new alternatively spliced FOXM1 variant FOXM1ΔC with an N-terminus and DNA-binding domain was reported recently in a variety of cancer cell lines.12 FOXM1 has been found to be aberrantly expressed in nearly all carcinomas.13 By controlling a panel of target genes involved in cell cycle progression, FOXM1 acts as a potent oncogene that induces mitosis and is thus considered a proliferation-specific transcription regulator.14, 15 Interestingly, recent studies have revealed the importance of FOXM1 in other cellular functions, including invasiveness and angiogenesis, by regulating the expression of matrix metalloproteinase-2/9 and vascular endothelial growth factor.16, 17 FOXM1 has also been shown to upregulate the expression of lysyl oxidase, ZEB1/2 and Slug, consequently leading to reduction the expression of E-cadherin.18, 19, 20, 21 Therefore, FOXM1 is suggested as an important regulator of EMT and metastasis.22 Despite these findings, the mechanisms by which different FOXM1 isoforms regulate tumor metastasis require further investigation.
Our further study on FOXM1D demonstrates that a novel isoform of FOXM1 can activate ROCKs by directly binding to these kinases. Moreover, overexpression of ectopic FOXM1D promoted marked cytoskeletal rearrangement and EMT, thus accelerating cancer invasion and metastasis. In colorectal cancer patients, FOXM1D expression significantly correlated with metastasis. Taken together, our results reveal FOXM1D as an important promoter of cancer cell metastasis via ROCKs activation and suggests that FOXM1D could be a potential biomarker or therapeutic target in colorectal cancer metastasis.
Results
Identification of FOXM1D in cancer cells
The potential isoforms of FOXM1 were screened utilizing in silico analysis. We further identified a novel transcript of FOXM1 by employing combined non-quantitative semi-nested reverse transcription PCR and GeneRacer PCR methods.23 In detail, complementary DNA was first obtained from diverse human cell lines using 5’ GeneRacer PCR to amplify only capped transcripts. The first-round PCR product was amplified using forward primer (FP) 1 in exon V and reverse primer (RP) 1 in exon VIII, whereas a second semi-nested PCR was performed with primers FP1 and RP2 in exon VIIa (Figure 1a). The indicated bands a, b, c and d in Figure 1b were extracted as the templates for additional semi-nested PCR. The results clearly showed that only bands c or d, but not a or b, could be used to amplify bands e or f, respectively, which represent two splice variants of FOXM1 (Figure 1c). Sequencing analysis demonstrated that band f originates from FOXM1 because of its incorporation of exon Va, whereas band e originates from a novel alternative splicing form owing to the absence of exon Va (Figure 1d). Then, FP2 that starts immediately from the translational initiating codon ATG, and RP3 that ends precisely with the terminal codon TAG, were designed to amplify this potential novel FOXM1 isoform (Figure 1a). Using PCR amplification followed by clone identification of T-vectors, we successfully isolated this novel FOXM1 isoform, which we termed FOXM1D owing to the absence of exon Va and presence of exon VIIa, and further verified it by sequencing analysis (Figure 1e and Supplementary Figure S1a).

Identification of FOXM1D. (a) Diagram of FOXM1 exons, the primers FP1, RP1 and RP2 for identifying FOXM1D, and the primers FP2 and RP3 for amplifying full-length FOXM1D. (b) Amplified fragments using the primers FP1 and RP1 in the first-round PCR. The arrows indicate the different amplified products (a–d) as templates in the second-round PCR. (c) The arrows indicate the amplified 533 and 488 bp PCR products corresponding to FOXM1D (e) and FOXM1A (f), which were amplified by the primers FP1 and RP2 in the second-round PCR. (d and e) Sequencing result (d) of band e in (c) indicates FOXM1D, which retains the VIIa exon that is absent in FOXM1B (e). (f–i) mRNA levels of FOXM1A (f), FOXM1B (g), FOXM1C (h) and FOXM1D (i) in human colorectal cancer cell lines as detected by qRT–PCR. (j) Diagram and sequences of the epitopes for polyclonal antibody preparation. CP11, joint region of exon V and VI; CP12, part of exon VIIa; DBD, DNA-binding domain; TAD, transactivation domain. (k) Protein level of FOXM1D in human colorectal cancer cell lines as detected by the customized polyclonal antibodies CP11 and CP12. The ratios of detected target proteins against actin control were analyzed and shown under each immunoblot image. The data are represented as mean±s.d., n=3. See also Supplementary Figure S1.
Then we measured FOXM1D mRNA levels in a set of colorectal and lung cancer cell lines using real-time PCR. FOXM1D was abundant in SW-620, LoVo and HCT-116 (Figure 1i) and in Calu-3 and NCI-H1650 (Supplementary Figure S1b and c). Interestingly, five FOXM1D high expression cell lines are prone to metastasis.24 Besides, we also detected FOXM1A/B/C mRNA level in colorectal cancer cell lines (Figures 1f and h). As FOXM1B/C are the main isoforms, which have been studied mostly recently,25, 26 FOXM1C mRNA level is generally not high in the six colon cancer cell lines. Therefore, we used FOXM1B as a control for FOXM1D functional exploration in the following study.
Next, to determine FOXM1B/D protein levels, we generated two polyclonal antibodies, CP11 and CP12 directed against epitopes encoded by exons V–VI and exon VIIa, respectively (Figure 1j). Therefore, CP11 and CP12 recognize FOXM1B/D or FOXM1A/D (Supplementary Figure S1a). We first verified the specificity of CP11 and CP12 antibodies utilizing commercial FOXM1 (K-19) antibody as control by IP (Supplementary Figure S1d). The immunoblotting (IB) assay of CP12 demonstrated FOXM1D is basically consistent with high FOXM1D mRNA levels in those five cell lines (Figure 1k, Supplementary Figure S1e).
FOXM1D promotes EMT and metastasis in vitro and in vivo
FOXM1 is known to have a critical role in cell cycle progression.13 Initially, we determined whether FOXM1D could function as a proliferation-driven oncoprotein (Supplementary Figure S2). As a result, we found that FOXM1D showed less-potent roles in promoting proliferation and tumorigenesis than does FOXM1B (Supplementary Result, Supplementary Figure S2). As the metastatic cancer cells highly express FOXM1D, we studied the role of FOXM1D in metastasis. First, we checked whether FOXM1D would affect EMT. According to FOXM1D expression results (Figures 1i and k), we chose low-expresser SW-480 and high-expresser LoVo cells for further study. We exogenously expressed FOXM1D into SW-480 cells, and knocked down FOXM1D expression in LoVo cells by lentiviral infection (Figures 2d and e lower left panels). Initially, we detected basal FOXM1A/B/C expression in SW-FOXM1D and LoVo-shFOXM1D cells to exclude the potential effects of other FOXM1 isoforms (Supplementary Figure S3a and b). Interestingly, E-cadherin staining was remarkably reduced in FOXM1D-overexpressing SW-480 cells compared with control cells (Figure 2a). On the contrary, mesenchymal markers (N-cadherin and vimentin) were significantly increased in FOXM1D-overexpressing SW-480 cells (Supplementary Figure S4a). Cell morphological conversion was observed concomitantly, as FOXM1D-overexpressing SW-480 cells displayed a more extended and elongated shape than did control cells (Figure 2a), which represents a feature of EMT.27 Our studies using FOXM1D-KD LoVo cells further supported this statement. In these cells, E-cadherin expression was markedly increased with tight junctions and tense shapes that could be restored partially by transient transfection of exogenous FOXM1D, meanwhile mesenchymal markers were reduced (Figure 2b, Supplementary Figure S4a). We further determined the FOXM1D-induced classical EMT by detecting Snail/slug/Twist1/2 and P-cadherin28 (data not shown). Moreover, FOXM1D overexpression increased both the migration and invasive capabilities of SW-480 cells, whereas FOXM1D-KD LoVo cells decreased the migration and invasive abilities, which were rescued by exogenously FOXM1D expression (Figure 2c, Supplementary Figure S4d–f). These results were repeatable in FOXM1D-overexpressing HeLa cells (Supplementary Figure S4b and c).

FOXM1D Promotes EMT and Metastasis in vitro and in vivo. (a and b) IF staining for E-cadherin (green) and nuclei (DAPI, blue) was performed in SW-480 cells transfected with control or FOXM1D-expressing (a) or in LoVo cells transfected with control, FOXM1D shRNA or FOXM1D shRNA with simultaneous FOXM1D-restored vector (b) (scale bar, 25 μm). FOXM1D sufficiency decreased and FOXM1D insufficiency increased E-cadherin level. The cell morphological changes between the FOXM1D-sufficient and -insufficient cells, as determined by microscopy, are shown in the right panel (scale bars, 200 μm and 40 μm, respectively). (c) A transwell assay determined that FOXM1D sufficiency promotes and FOXM1D insufficiency inhibits the invasion capability of the indicated cells. The quantified results are shown in Supplementary Figure S4f. (d–e) FOXM1D sufficiency promotes and FOXM1D insufficiency impairs cancer progression and metastasis. SW-480 cells transfected with the control vector or with FOXM1D-expressing vector (d, upper panel) or LoVo cells transfected with shRNA-control or shRNA-FOXM1D (e, upper panel) were implanted in the foot pad or orthotopic cecum of BALB/c nude mice, respectively. The metastasis incidence and signal intensities in each group are shown at the bottom. The red arrows indicate orthotopic tumor formation and metastatic liver nodules (d and e, bottom right). The efficacy of overexpressing or knocking down FOXM1D was determined by IB assay (d and e, bottom left). The images are representative of at least seven implanted mice. **P<0.01 vs control groups. See also Supplementary Figures S2.
To further verify the role of FOXM1D, we constructed FOXM1A/B/C-overexpressing SW-480 cells as controls and detected the EMT related markers and metastatic capabilities in vitro. Immunofluorescence (IF) and immunoblot assays showed that E-cadherin levels decreased remarkably in FOXM1C/D-overexpressing cells and moderately in FOXM1B-overexpressing cells but not in FOXM1A-overexpressing SW-480 cells (Supplementary Figure S5a and b). Interestingly, the expression level of N-cadherin significantly increased only in FOXM1D-overexpressing SW-480 cells (Supplementary Figure S5a, middle). Moreover, both FOXM1B/C-overexpressing cells remarkably increased the migration and invasion comparing to control cells. However, FOXM1D displayed the most potency in promoting migration and invasion compared with FOXM1A/B/C (Supplementary Figure S5c–e). Intriguingly, we observed the obvious cell morphological alteration only in FOXM1D-overexpressing SW-480 cells with an elongated and dissociated shape indicating different mechanisms involved in FOXM1D-induced EMT and metastasis.
In in vivo study, we used both foot fat pad injection and orthotropic xenograft models to investigate the metastasis-promoting potential of FOXM1D.29 In the foot fat pad model, we observed that FOXM1D-overexpressing SW-480 cells remarkably promotes inguinal lymph node metastases (7/7) comparing with control (1/7) (Figure 2d, upper left). In contrast, FOXM1D knockdown in LoVo cells significantly reduced metastases (0/7) than control (6/7) (Figure 2e, upper left). In the orthotopic xenograft model, mice implanted with FOXM1D-overexpressing SW-480 cells suffered from multiple metastases in the liver and abdominal cavity (7/8) compared with control (2/7) (Figure 2d, upper right). FOXM1D knockdown in LoVo cells reduced metastasis markedly (1/8) versus control (6/8) (Figure 2e, upper right). The signal intensities of lymph node and distant metastases were both quantified using the Bruker MI software (Billerica, MA, USA; Supplementary Figure S4g and h). Taken together, these results demonstrate that FOXM1D can promote colorectal cancer cell EMT and metastasis.
High FOXM1D expression correlated with colorectal cancer metastases in FUSCC cohort
Given that FOXM1D promotes cancer cell metastasis in our mouse xenografted tumor model systems (Figures 2d and e), we further examined the expression of FOXM1D in CRC samples of FUSCC cohort. Seventeen patients would be sufficient for the clinical samples (power 80%, α=0.05). There were no statistical difference in clinicopathological parameters between 24 non-metastatic and 22 metastatic CRC patients (Supplementary table S1). The primary tumors were examined by quantitative RT–PCR. FOXM1D was not expressed differently between tumor and paired normal tissues in non-metastatic CRC samples (Supplementary Figure S6a). However, FOXM1D exhibited a marked increase in the primary tumors of metastatic groups (P<0.001) (Supplementary Figure S6b). Moreover, we also detected the expression of FOXM1A/B/C in CRC samples. FOXM1A demonstrated no difference in both two groups samples (Supplementary Figure S6c and d). The expression of FOXM1B/C showed highly significant differences in non-metastatic tumor and paired normal tissues (P<0.001) (Supplementary Figure S6e and g). In addition, the expression of FOXM1B increased in metastatic tumor group (P<0.05) and FOXM1C did not (Supplementary Figure S6f and h). However, the expression of FOXM1D was much higher than FOXM1B in metastatic tumor group in consideration of primer efficiency (data not shown). Altogether, the expression of FOXM1D correlated with colorectal cancer metastasis.
FOXM1D interacts directly with ROCKs
In mechanical study, we performed co-immunoprecipitation (co-IP)-coupled mass spectrometric analysis and identified ROCK2 as one main FOXM1D-binding protein (Figure 3a and Supplementary Figure S7a). Next, the physical interaction between FOXM1D and ROCK2 was validated using a glutathione S-transferase (GST) pull-down assay in both LoVo and SW-480-FOXM1D cells (Figures 3b and c) as well as by co-IP-IB assays in 293 T cells that overexpressed Flag-tagged FOXM1D (Figure 3d). This result was reproduced by reverse co-IP-IB assays using the anti-ROCK2 antibody (Figure 3e). More convincingly, this result was further confirmed by a double co-IP with CP11 and CP12 antibodies, respectively, excluding the possible interference of this interaction by endogenous FOXM1A (Figures 3f and g). Also, we observed that FOXM1D mainly resides in the cytoplasm (Supplementary Figure S7b). Our IF assays showed that ROCK2 is colocalized with FOXM1D, but not FOXM1A/B/C, in the cytoplasm of 293 cells (Figure 3h and Supplementary Figure S7c). Consistently, ROCK2 did not interact with either of the FOXM1A, 1B and 1C proteins by co-IP-IB assays (Supplementary Figure S7d). Therefore, these results demonstrate that only FOXM1D interacted with ROCK2. Like ROCK2, ROCK1 also bound to FOXM1D as confirmed by using GST pull-down and co-IP assays (Figures 3i and j), suggesting that FOXM1D may influence EMT and metastasis by interacting with both of ROCK1 and ROCK2.

FOXM1D Directly Interacts with ROCKs. (a) ROCK2 was identified as one of FOXM1D-binding proteins. 293 T cells were transfected with Flag-tagged FOXM1D-expressing or control plasmid. IP was performed with the cell extracts using anti-Flag antibody. Subsequently, the IP complex was subjected to LC-MS/MS analysis. (b and c) GST-FOXM1D binds to ROCK2. GST pull-down was performed using GST-FOXM1D and total cell extracts of LoVo (b) or SW-480 (c) cells, followed by IB with anti-ROCK2 antibody. (d) Flag-tagged FOXM1D binds to ROCK2. 293 T cells were transfected with Flag-FOXM1D-expressing or control plasmid, followed by IP using anti-Flag antibody and IB with anti-ROCK2 antibody. (e) Reciprocal IP was performed in the indicated cells using anti-ROCK2 antibody, followed by IB with CP11 and CP12 antibodies. (f) Schematic diagram of Re-IP, that is, two-step IP using CP11 and CP12 antibodies. (g) Two round co-IP was performed using the indicated cell extracts. A greater amount of ROCK2 was captured in SW-480-FOXM1D cells. (h) FOXM1D colocalizes with ROCK2. Triple IF staining for Flag-FOXM1D (green), HA-ROCK2 (red), and nuclei (DAPI, blue) was performed in 293 cells that were transiently co-transfected with pFLAG-CMV-4-FOXM1D and pCMV-C-HA-ROCK2. Scale bar, 25 μm. (i) GST-FOXM1D also binds to ROCK1. GST pull-down was performed using GST-FOXM1D and LoVo total cell extracts, followed by IB with anti-ROCK1 antibody. (j) Flag-tagged FOXM1D also binds to ROCK1. 293 T cells were transfected with Flag-tagged FOXM1D-expressing or control plasmid, followed by IP using anti-Flag and IB with anti-ROCK1 antibody. See also Supplementary Figure S7.
FOXM1D is crucial for the ROCKs ability to regulate actin arrangement and EMT in a Rho-dependent manner
Because ROCKs have an important role in cytoskeletal rearrangement and EMT by inducing the phosphorylation of several target substrates,5, 6 we examined the actin organization in proficient and deficient cells. The fluorescence intensities of F-actin and phospho-myosin light chain (MLC) in the cytoplasm were quantified. In FOXM1D-overexpressing SW-480 cells, actin polymerization increased as shown by phalloidin staining of F-actin compared with control SW-480 cells (Figures 4a and b). As an essential substrate of ROCKs, MLC phosphorylation also increased significantly in the cytoplasm of these cells (Figure 4a), which subsequently enhances actomyosin contractility. By striking contrast, F-actin was depolymerized in FOXM1D knockdown cells, compared with control LoVo cells, which was rescued by overexpression of ectopic FOXM1D (Figure 4b). Interestingly, both F-actin and phospho-MLC in FOXM1D knockdown LoVo cells moved near the cell membrane from the cytoplasm (Figure 4b).

FOXM1D is crucial for the ROCKs’ ability to regulate actin arrangement and EMT in a Rho-dependent manner. (a–b) Triple IF staining for F-actin (phalloidin, green), phospho-MLC (red) and nuclei (DAPI, blue) in SW-480 cells transfected with control and FOXM1D-expressing vector (a) or in LoVo cells transfected with shRNA-control, shRNA-FOXM1D or shRNA-FOXM1D and restored FOXM1D-expressing vector (b). The cell morphological changes and F-actin assembly among these cells are shown. Scale bar, 25 μm. (c) IB assay. The expression and/or phosphorylation of ROCKs downstream targets in the indicated cells. (d–e) Triple IF staining for F-actin (phalloidin, green), phospho-MLC (red) and nuclei (DAPI, blue) was performed to detect cytoskeletal changes. SW-480-FOXM1D cells were treated with Rho inhibitor (1 μg/ml) for 4 h (d). LoVo-shRNA-FOXM1D cells were treated with Rho activator (1 U/ml) for 30 min (e). Scale bar, 25 μm. (f) IB assay. The expression and/or phosphorylation of E-cadherin, ROCKs downstream targets in the indicated cells treated with Rho inhibitor or Rho activator. (g) ROCKs activity was measured by ELISAs. In SW-480 cells, FOXM1D overexpression enhanced ROCKs activity, which was inhibited by Rho inhibitor. In LoVo cells, FOXM1D knockdown decreased ROCKs activity, and Rho activator treatment induced a slightly increase in ROCKs activity in FOXM1D knockdown cells. Restored FOXM1D expression recovered ROCKs activity. The data are represented as mean±s.d. for triplicate samples. *P<0.05 and **P<0.01. See also Supplementary Figure S8.
Correlated with the morphological phenotypic changes as described above, the phosphorylation, but not expression, levels of LIMK1/2, cofilin and MLC (involved in actomyosin contractility) were increased markedly in FOXM1D-overexpressing SW-480 cells (Figure 4c, left panel), but reduced markedly in FOXM1D-deficient LoVo cells (Figure 4c, right panel). The phosphorylation levels of LIMK1/2, cofilin and MLC phospho-cofilin was restored in FOXM1D-deficient LoVo cells by ectopically expressed FOXM1D (Figure 4c, right panel). These results indicate that the activity of ROCKs is influenced by FOXM1D. Further, the ROCK activity as measured by enzyme-linked immunosorbent assay was enhanced significantly in FOXM1D-overexpressing SW-480 cells (Figure 4g), but decreased considerably in FOXM1D knockdown LoVo cells (Figure 4g).30 We also repeated the experiments in HeLa cells (Supplementary Figure S8c).
Next, we determined whether ROCKs activation by FOXM1D is Rho-dependent, as ROCKs can be activated by arachidonic acid and granzyme B in a Rho-independent manner.31, 32 We observed the morphological changes of SW-480-FOXM1D cells treated with Rho inhibitor I. The cell size decreased notably, along with F-actin and phospho-MLC translocation from the cytoplasm to the membrane (Figure 4d). The expression of E-cadherin increased, and the phosphorylation of LIMK1/2, MLC and cofilin significantly decreased, which was consistent with decreased ROCKs activity as determined by enzyme-linked immunosorbent assay (Figure 4f, left panel, Figure 4g and Supplementary Figure S8d). In addition, we found that the cell size, stress fiber formation and F-actin levels of LoVo control cells increased remarkably after treatment of control LoVo cells with Rho activator I, indicating its strong potency (Supplementary Figure S8a). Subsequent results showed that activation of ROCKs by Rho agonist was FOXM1D-dependent (Figures 4e and f, right panel and Supplementary Figure S8b). Taken together, these results indicate that both FOXM1D and Rho-GTPases are interdependently required for ROCKs activation, actin polymerization and cytoskeletal rearrangement.
ROCKs inhibitors abrogate FOXM1D-induced ROCKs activation
As two specific inhibitors of ROCKs, Y-27632 and fasudil, have been shown to reduce tumor progression in various types of cancers,8 we tested whether Y-27632 and fasudil could also abolish FOXM1D-induced ROCKs activation by IF assays. These two inhibitors induced depolymerized actin, remarkably reduced phospho-MLC (Ser19) levels and altered cell shape of FOXM1D-overexpressing SW-480 and LoVo cells (Figure 5a and Supplementary Figure S9a), compared with that in control SW-480 or LoVo cells (Figures 4a and b). They also induced E-cadherin expression (Figure 5b and Supplementary Figure S9b) as well as reduced phosphorylation of ROCKs’ substrates in FOXM1D-overexpressing SW-480 and LoVo cells (Figure 5d and Supplementary Figure S9d) and ROCK activity as measured by enzyme-linked immunosorbent assay in both FOXM1D-overexpressing SW-480 and LoVo cells (Figure 5c and Supplementary Figure S9c). These results suggest that FOXM1D-induced ROCKs activation may contribute to destruction of cell junctions. It may result from the reduced expression of E-cadherin and phosphorylation of other ROCKs-targeted substrates. The destruction of cell junctions could be reversed by Y-27632 or fasudil. As Y-27632 administration did not interfere with the binding of FOXM1D to ROCK2 (Figure 5e), Y-27632 may bind to ROCK2 at a site distinct from that of FOXM1D, suggesting that this inhibitor may abolish FOXM1D activation of ROCKs activation by an allosteric mechanism.

ROCKs Inhibitors Abrogate FOXM1D-Induced ROCKs Activation. (a–b) Triple IF staining for F-actin (phalloidin, green), phospho-MLC (a) or E-cadherin (b) (red) and nuclei (DAPI, blue) showed decreased F-actin and phosphorylated MLC, increased E-cadherin and altered cell shape and size in SW-480-FOXM1D cells treated with the ROCKs inhibitor Y-27632 or fasudil (both 10 μM, for 24 h). Scale bar, 25 μm. (c) Y-27632 significantly reduced ROCKs activity in SW-480-FOXM1D cells as measured by ELISAs. The data are represented as mean±s.d. for triplicate samples. * P<0.05. (d) IB assay. The expression and/or phosphorylation of E-cadherin and ROCKs downstream targets in SW-480-FOXM1D cells treated with Y-27632. (e) Y-27632 does not affect the binding of FOXM1D to ROCK2, indicating that the binding site of FOXM1D differs from that of Y-27632 on ROCK2. SW-480-FOXM1D cells were treated with Y-27632 (10 μM, for 24 h), and then, IP was performed using anti-ROCK2 antibody, followed by IB with CP12 antibody. See also Supplementary Figure S9.
Mapping of the binding domains of FOXM1D and ROCK2
To further interrogate the mechanism for FOXM1D activation of ROCK2, we tried to define the binding domains of FOXM1D and ROCK2. ROCK2 consists of an N-terminal kinase domain, a potential coiled-coil-forming region that contains a Rho-binding domain (RBD), and a C-terminal domain with an auto-inhibitory capability of blocking its own kinase activity (Figure 6A (ai).5 We generated a series of hemagglutinin-tagged ROCK2 truncated mutants as shown in Figure 6A (ai) and expressed in 293 T cells (marked asterisk in Figure 6B (bi). Using these constructs, we conducted transient transfection followed by co-IP assays and found that only hemagglutinin antibody bound intact ROCK2 (a) and three ROCK2 mutants (c, d and e) could capture FOXM1D (Figure 6B (bi), upper panel). Furthermore, when Flag-tagged FOXM1D was co-expressed in the above 293 T cells, the IF assay yielded results that were consistent with the co-IP results, that is, ROCK2 mutants b and f failed to bind to FOXM1D (Figure 6D). Thus, the RBD-containing coiled-coil region (residues 686–1145) of ROCK2 interacts with FOXM1D. Next, using co-IP assay we defined the precise FOXM1D-binding domain (FBD) in ROCK2 by further chopping the RBD-containing coiled-coil region into three truncated mutants (Figure 6A (aii)). The result shown in Figure 6B (bii) determined that mutant g (residues 686–824) adjacent to RBD was the minimal essential FBD. Meanwhile, the result of GST pull-down assay further verified the interaction between GST-FOXM1D and ROCK2 (686–824 aa), that is, mutant g (Figure 6B (biii)). The coiled-coil usually contains a heptad repeat of hydrophobic and charged amino-acid residues, representing a highly versatile functional motif.33, 34 We screened the residues 686–824 region of ROCK2, and then mutated the seven hydrophobic I/L to hydrophilic G (Figure 6C (ci)). Among the four ROCK2 coiled-coil mutants, the first and second mutants totally abolished, and the third and fourth mutants significantly impaired the binding activity of ROCK2-FBD region to FOXM1D (Figure 6C (cii)). Therefore, these results indicate that the coiled-coil motif with a heptad repeat of hydrophobic and charged amino-acid residues in residues 686–824 region of ROCK2 is an essential unit for the intact binding of ROCK2 to FOXM1D.

Identification of interacting sites of FOXM1D and ROCK2. (A) Schematic illustration of truncated mutants of ROCK2 (Aai) and its coiled-coil region (686–1145 aa) (Aaii). (B) A series of HA-tagged ROCK2 and its truncated mutants (mutants A–F in figure ai, and mutants G-I in figure aii, all of which were marked by asterisks in figure bi and bii) were expressed in 293 T cells and then subjected to IP using anti-HA antibody, followed by IB with CP12 antibody. In (Bbi), only intact ROCK2 (A) and three mutants (C–E) captured FOXM1D, and further, only mutant g captured FOXM1D using IP and GST pull-down assay, respectively (Bbii and Bbiii). All of these constructs are marked as ‘+’ in (A). (C) Schematic illustration of minimal FOXM1D-binding domain and its site-mutated mutants in (Cci). The mutants were expressed in 293 T cells and subjected to IP using anti-HA antibody as previously mentioned. Mutants 3 and 4 showed a subtle binding with FOXM1D in (Ccii). (D) Triple IF staining for HA-ROCK2 deletion mutants (red), Flag-FOXM1D (green) and nuclei (DAPI, blue) showed the cellular colocalization between FOXM1D and the ROCK2 truncated mutants indicated in (bi). Scale bar, 25 μm. (E) Schematic illustration of FOXM1D and its truncated mutants. (F) A series of Flag-tagged FOXM1D and its truncated mutants (A–E marked by asterisks) were expressed in 293 T cells and then subjected to IP using anti-Flag antibody, followed by IB with ROCK2 antibody. Only intact FOXM1D (A) and its mutants (B, C and possibly D) captured ROCK2; these constructs are marked as ‘+’ in (E). (G) ROCKs activity in 293 T cells that expressed intact FOXM1D or its mutants was measured by ELISAs. The data are represented as mean±s.d. for triplicate samples. *P<0.05 vs blank. See also Supplementary Figure S10.
To map the ROCK2-finding domain in FOXM1D, we generated five FOXM1D mutants (named a–e) (Figure 6E) and expressed them in 293 cells (Figure 6F). The co-IP experiment showed that only those FOXM1D fragments (a, b, c and d) containing residues 218–406 (encoded by exons IV, V and VI) bound to ROCK2 (Figure 6F), indicating that this region is the essential region for FOXM1D to physically bind to ROCK2. Although FOXM1A/B/C contain fragments of residues 218–406, the other parts might conformationally shield this region owing to steric hindrance, leading to the inability to bind to ROCKs. Consistently, only FOXM1D fragments a, b and c, which contain residues 218–454 with exon VIIa, could activate ROCKs activity after transfection of 293 cells (Figure 6G), suggesting the critical role of the insertion of an additional exon VIIa in FOXM1D-dependent activation of ROCKs activity. We further tested whether the addition of exon Va would have an impact on the interaction between FOXM1D and ROCK2. The 218–469 region (contains both exon Va and VIIa) of FOXM1A were constructed into the Flag-tag plasmid. The IP assay demonstrated that 218–469 region of FOXM1A failed to bind to ROCK2 comparing to full-length FOXM1D, indicating that the existence of exon Va may induce steric hindrance for the interaction with ROCK2 (Supplementary Figure S10). Therefore, these results strongly indicate that the region of FOXM1D, containing exon VIIa, is essential for binding to the FBD of ROCK2, and this binding is important for ROCK2 activation.
Colocalization of FOXM1D and ROCK2 in clinical samples
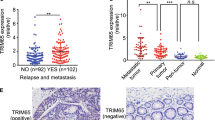
At last, we employed the immunohistochemistry (IHC) method to detect the expression of FOXM1D using CP12 in distinct colorectal tumor or normal tissues. To avoid FOXM1A interference, we compared the expression levels of FOXM1A and FOXM1D in five non-metastatic and five metastatic tumor tissues using quantitative RT–PCR. Consistent with the previous data, FOXM1A was negligibly expressed in both types of tissues compared with FOXM1D and beta-actin (Supplementary Figure S11). Thus, CP12 staining principally represents FOXM1D expression. The IHC results revealed that FOXM1D is highly expressed in primary and metastatic liver tissues of 22 colorectal cancer patient, but not in 24 non-metastatic colorectal cancer samples and paired normal colorectal samples (a representative result is shown in Supplementary Figure S12a, left panel). Correspondingly, ROCK2 was also highly expressed in metastatic, but not non-metastatic, colorectal tumors (Supplementary Figure S12a, middle panel). In addition, the IHC score assessment revealed that FOXM1D expression in metastatic samples was markedly higher than that in non-metastatic samples (P<0.001) (Supplementary Figure S12b). Their expressions were correlated with each other in these tumor samples (Supplementary Figure S12c). Therefore, these findings demonstrate that FOXM1D is associated with colorectal cancer metastasis, suggesting that FOXM1D might be a potential biomarker for progressive colorectal cancer.
Discussion
FOXM1 has been generally regarded as a nuclear proliferation-specific transcription factor that is essential for regulation of the cell cycle, specifically for timely entry into both the S phase and mitosis.14, 15 Consistently, FOXM1 has also been shown to function as a potent oncoprotein that participates in a series of tumorigenesis stages, from neoplasia to distant metastasis.35 Despite the existence of a nuclear localization sequence, nuclear localization of FOXM1C requires phosphorylation by Raf/MEK/MAPK signaling.36 Interestingly, Wnt3a increases the level and nuclear accumulation of FOXM1 (B), which subsequently mediates β-catenin nuclear translocation and promotes glioma tumorigenesis.37 Regardless of these studies, it remains unclear whether and how exactly this family regulates cancer metastasis. The studies presented here not only identified FOXM1D as a novel member of this family, but also demonstrated its role in promoting EMT and colorectal cancer cell metastasis.
Unlike other isoforms (FOXM1B/C) that promote metastasis by regulating the expression (for example, matrix metalloproteinases, vascular endothelial growth factor, lysyl oxidase, slug and stathmin) or location (β-catenin) of the associated molecules, FOXM1D employs a unique mechanism to promoting EMT and metastasis by activating ROCKs together with Rho-GTPases, thus leading to cell detachment, motility and invasion by cytoskeletal rearrangement.
Even there is only a difference in a few dozen amino acids among the four FOXM1 isoforms, FOXM1D exerts completely distinct functions and cellular localization compared with other FOXM1 isoforms. Therefore, cancer cells may take advantage of the alternative splicing and auto-regulation loop to produce different FOXM1 isoforms that can help the cells advance to more aggressive stages.38, 39
Our studies further showed that FOXM1D promotes cancer cell metastasis by interplaying with Rho/ROCK pathway. The Rho GTPase family members are best known as key regulators of various cell biological processes, including cell shape, motility, proliferation and apoptosis, by regulating actin cytoskeleton organization and dynamics.5 Two ROCK isoforms, ROCK1 and ROCK2, exist in human cells and share 65% sequence identity overall and 92% identity in their kinase domains. ROCKs are activated when GTP-bound RhoA/C bind to RBD in the coiled-coil region of ROCKs, converting the ROCK kinase from a closed auto-inhibitory conformation to an open conformation. Therefore, RhoA/C family members promote actomyosin contractile force generation via the ROCK1/2-mediated phosphorylation of numerous downstream target proteins, including LIMK1/2, MLC and the myosin-binding subunit of MLC phosphatase, to inhibit catalytic activity and subsequent MLC dephosphorylation. These events contribute directly to several proximal processes, including the regulation of morphology, motility, and cell–cell and cell–matrix adhesion. Our studies as described here add another novel player in this regulation. We showed that FOXM1D is crucial for ROCKs activation. As shown in Figure 7, FOXM1D bound to the FBD of ROCKs via its specific fragment encoded by exons V, VI, VII and VIIa, whereas GTP-bound Rho bound directly to the RBD adjacent to FBD. The assembly of the heterotrimer releases ROCKs activity from an auto-inhibitory status. Accordingly, the activated ROCKs induce the phosphorylation of a series of substrates, thus leading to F-actin assembly and actomyosin contraction, which are prerequisites for cancer cell EMT and metastasis.

Schematic diagram of the role of FOXM1D in ROCKs activation. The simultaneous binding of FOXM1D to FBD and active Rho-GTP to RBD in the ROCKs coiled-coil region activates ROCKs. ROCKs activation impairs E-cadherin level and phosphorylates LIMK1/2, cofilin and MLC that regulate F-actin rearrangement and cell shape collaboratively, eventually leading to EMT and metastasis. PH, pleckstrin homology domain; CRD, cysteine-rich domain; RBD, Rho-binding domain (ROCK2 residues 934–1015); FBD, FOXM1D-binding domain (ROCK2 residues 686–824), which interacts with the FOXM1D region coded by exons V, VI, VII, VIIa.
We also demonstrate that the sharp changes are accompanied by cytoskeletal remodeling in FOXM1D overexpression or knockdown cancer cells. ROCKs inactivation by RhoA/C knockdown induces a notable increase in E-cadherin expression.34 Furthermore, ROCKs inactivation by Rho or ROCKs inhibitors or FOXM1D knockdown produces a significant increase in E-cadherin expression. Moreover, ROCKs activation may induce additional distal cellular effects, such as gene transcription, proliferation, differentiation, apoptosis and oncogenic transformation,40 further highlighting the significance of FOXM1D in tumor progression.
Consistent with the above findings, the pharmacologic inhibitors of ROCKs, Y-27632 and fasudil, which target the ATP-dependent kinase domain, inhibit ROCK1 and ROCK2 at equimolar concentrations. Fasudil has been approved for clinical application in China and Japan since 1995. This inhibitor was shown to inhibit tumor dissemination and metastasis.41 Moreover, Y-27632 displays potent anti-tumor invasion and metastasis activity in mice.42, 43 Considering that the binding site of FOXM1D is distinct from those of Y-27632 and fasudil in ROCKs, these ROCKs inhibitors may inactivate FOXM1D-induced ROCKs activation through an allosteric mechanism.
In summary, our studies demonstrate FOXM1D as a novel isoform of FOXM1 and a new player in cancer cell EMT and metastasis by directly binding to ROCKs and activating their kinase activity. Also, we showed the high correlation of the FOXM1D expression level with metastasis in colorectal cancer patients. This finding suggests that FOXM1D might be a prospective biomarker for metastasis evaluation and a potential target for future development of anti-metastatic cancer therapy.
Materials and methods
Study approval
Animal studies were approved by the Animal Ethics Committee at Shanghai Medical School, Fudan University. Forty-six fresh-frozen colorectal cancer samples and 24 adjacent normal colon tissues were obtained from the Tissue Bank at Fudan University Shanghai Cancer Center. All patients signed written consent forms, and the use of these samples was approved by the Ethics Committee at Fudan University Shanghai Cancer Center.
GeneRacer PCR for FOXMD identification
GeneRacer PCRs were performed according to the manufacturer’s protocol (Invitrogen, Hudson, NH, USA). The first PCR amplification was performed using the complementary DNA obtained by 5’ GeneRacer PCR with the primers FP1 and RP1 (Figure 1a). The first-round PCR product was extracted to perform nested PCR with the primers FP1 and RP2. The nested PCR product was analyzed on an agarose gel. Finally, the bands were excised from the gel, cloned into a pMD-19 T-vector (Takara Bio, Dalian, China), and sequenced. The coding sequences of FOXM1A/B/C were also cloned for future study by PCR using the primer pairs FR2 and RP3 (primer sequences are listed in Supplementary Table S2).
In vivo metastatic animal models and bioluminescent imaging
The metastatic animal study was performed by animal experiment technicians for blinding. Male BALB/c nude mice (5 weeks) were purchased from Shanghai SLAC Animal Laboratory of the Chinese Academy of Sciences. The animals were marked and allocated randomly. For the lymph node metastasis assays, 1.5 × 106 of the cells shown in the left panel of Figures 2d and e were suspended in 100 μl of serum-free media and rear injected into the hind limb foot pad (seven mice/group). To detect metastases of orthotopic colorectal cancer, 3 × 106 of the cells shown in the right panel of Figures 2d and e were suspended in 100 μl of serum-free media and injected subcutaneously into nude mice. After 2 weeks, the subcutaneous tumors were collected sterilely and diced into 1 mm3 pieces. Before transplantation, the cecal wall was damaged slightly to facilitate tumor cell infiltration. Then, the small tumor cubes were seeded on the blind end of the cecum of the nude mice (eight mice/group). For the in vivo metastatic animal study, the power value of each group was enough to detect the effect size (power 92.3%, α=0.05). In vivo tumor formation and metastasis were tracked by bioluminescence. Before imaging, d-luciferin (PerkinElmer, Waltham, MA, USA) was injected intraperitoneally into the mice (150 mg/kg). After 5 min, bioluminescence was performed using an In Vivo system (In Vivo MS FX PRO, Bruker). Luminescent images were captured with 300 s of exposure time. The signal intensities of the metastasis sites were measured by Bruker MI software. The resulting images were automatically merged with X-ray and luminescent photographs using the In Vivo system software. The mice were euthanized and dissected after 8 weeks.
Statistics
Sample size estimation and power calculation were determined by PASS 11.0 (NCSS, Kaysville, UT, USA). The significance of the in vivo metastatic assays (foot pad and orthotopic) was determined by Fisher’s exact test. Student’s t-test was used in continuous variable with similar variance. In detail, the significance of ROCKs activity, the quantitative PCR results from patient specimens, the signal intensities of metastasis sites, and the in vitro assays was determined by Student’s t-test (two-tailed). Multiple group variables with similar variance were performed by one-way analysis of variance. The significance of IHC staining scores of ROCK2 and FOXM1D was determined by an independent samples t-test using SPSS software. The significance of clinical characteristics of colorectal patients was determined by Student’s t-test or χ2-test. The correlation of IHC staining scores for ROCK2 and FOXM1D was determined by Pearson’s correlation test (two-tailed). *P<0.05 was considered significant. **P<0.01 was considered remarkably different. ***P<0.001 was considered highly remarkably different.
References
Siegel R, Ma J, Zou Z, Jemal A . Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29.
Liu X, Ji Q, Fan Z, Li Q . Cellular signaling pathways implicated in metastasis of colorectal cancer and the associated targeted agents. Future Oncol 2015; 11: 2911–2922.
Chaffer CL, Weinberg RA . A perspective on cancer cell metastasis. Science 2011; 331: 1559–1564.
Etienne-Manneville S, Hall A . Rho GTPases in cell biology. Nature 2002; 420: 629–635.
Riento K, Ridley AJ . Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 2003; 4: 446–456.
Lamouille S, Xu J, Derynck R . Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–196.
Shi JW, Liu W, Zhang TT, Wang SC, Lin XL, Li J et al. The enforced expression of c-Myc in pig fibroblasts triggers mesenchymal-epithelial transition (MET) via F-actin reorganization and RhoA/Rock pathway inactivation. Cell Cycle 2013; 12: 1119–1127.
Morgan-Fisher M, Wewer UM, Yoneda A . Regulation of ROCK activity in cancer. J Histochem Cytochem 2013; 61: 185–198.
Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S . An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med 1999; 5: 221–225.
Kalin TV, Ustiyan V, Kalinichenko VV . Multiple faces of FoxM1 transcription factor. Cell Cycle 2014; 10: 396–405.
Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol 1997; 17: 1626–1641.
Kim YH, Choi MH, Kim JH, Lim IK, Park TJ . C-terminus-deleted FoxM1 is expressed in cancer cell lines and induces chromosome instability. Carcinogenesis 2013; 34: 1907–1917.
Wierstra I . FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv Cancer Res 2013; 119: 191–419.
Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol 2005; 25: 10875–10894.
Myatt SS, Lam EW . The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007; 7: 847–859.
Zhang Y, Zhang N, Dai B, Liu M, Sawaya R, Xie K et al. FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res 2008; 68: 8733–8742.
Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH . Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res 2007; 67: 8293–8300.
Park HJ, Gusarova G, Wang Z, Carr JR, Li J, Kim K-H et al. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med 2011; 3: 21–34.
Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-beta-dependent cancer metastasis. J Clin Invest 2014; 124: 564–579.
Yang C, Chen H, Tan G, Gao W, Cheng L, Jiang X et al. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett 2013; 340: 104–112.
Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A et al. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem 2011; 112: 2296–2306.
Raychaudhuri P, Park HJ . FoxM1: a master regulator of tumor metastasis. Cancer Res 2011; 71: 4329–4333.
Rohaly G, Chemnitz J, Dehde S, Nunez AM, Heukeshoven J, Deppert W et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell 2005; 122: 21–32.
Rajput A, Dominguez San Martin I, Rose R, Beko A, Levea C, Sharratt E et al. Characterization of HCT116 human colon cancer cells in an orthotopic model. J Surg Res 2008; 147: 276–281.
Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res 2006; 66: 3593–3602.
Teh MT . FOXM1 coming of age: time for translation into clinical benefits? Front Oncol 2012; 2: 146.
Lee JM, Dedhar S, Kalluri R, Thompson EW . The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172: 973–981.
Sun L, Hu H, Peng L, Zhou Z, Zhao X, Pan J et al. P-cadherin promotes liver metastasis and is associated with poor prognosis in colon cancer. Am J Pathol 2011; 179: 380–390.
Tseng W, Leong X, Engleman E . Orthotopic mouse model of colorectal cancer. J Vis Exp 2007, e-pub ahead of print 4 December 2007 doi:10.3791/484.
Liu PY, Liao JK . A method for measuring Rho kinase activity in tissues and cells. Methods Enzymol 2008; 439: 181–189.
Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N et al. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem 1999; 274: 3744–3752.
Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J . Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J Exp Med 2005; 201: 465–471.
Burkhard P, Stetefeld J, Strelkov SV . Coiled coils: a highly versatile protein folding motif. Trends Cell Biol 2001; 11: 82–88.
Parry DA, Fraser RD, Squire JM . Fifty years of coiled-coils and alpha-helical bundles: a close relationship between sequence and structure. J Struct Biol 2008; 163: 258–269.
Halasi M, Gartel AL . FOX(M1) news–it is cancer. Mol Cancer Ther 2013; 12: 245–254.
Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY, Yao KM . Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell ScI 2005; 118: 795–806.
Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011; 20: 427–442.
Halasi M, Gartel AL . A novel mode of FoxM1 regulation: positive auto-regulatory loop. Cell Cycle 2009; 8: 1966–1967.
Zhang J, Manley JL . Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 2013; 3: 1228–1237.
Rath N, Olson MF . Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep 2012; 13: 900–908.
Ying H, Biroc SL, Li WW, Alicke B, Xuan JA, Pagila R et al. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther 2006; 5: 2158–2164.
Itoh KYK, Akedo H, Uehata M, Ishizaki T, Narumiya S . An essential part for Rho associated kinase in the transcellular invasion of tumor cells. Nat Med 1999; 5: 221–225.
Wang Y, Lei R, Zhuang X, Zhang N, Pan H, Li G et al. DLC1-dependent parathyroid hormone-like hormone inhibition suppresses breast cancer bone metastasis. J Clin Investig 2014; 124: 1646–1659.
Acknowledgements
This research was supported by the Major State Basic Research Development Program of China (2013CB910802), the National Natural Science Foundation of China (81171910, 81372258) and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning to WH. We thank Professor Thomas M Roberts (DFCI, Harvard Medical School) and Professor Hua Lu (Department of Biochemistry and Molecular Biology, Tulane University) for scientific suggestions and critical reading of this manuscript. We are also grateful to Dr Yuhu Xin for assistance with confocal microscopy; to Mrs Ping Zhang for assistance with flow cytometry; to Dr Yi Qin, who provided the pCMV-C-FLAG and pCMV-C-HA plasmids; to Professor Yanhui Xu, who kindly provided the modified pGEX-6P-1 plasmid; and to Professor Shenglin Huang, who provided the p-pLVX-IRES-Neo and pwpxl-eGFP-2A-CBGr99 lentiviral vectors; to Dr Fangning Wan for advices on statistics and revision of manuscript.
Author contributions
XZ designed, performed and analyzed the majority of the experiments and co-wrote the manuscript. LZ, YD, PZ, YS, JC, PD, NW, CY and QQ performed or repeated several experiments. HG provided technical support for IHC. TH analyzed the clinical data. HZ and YW processed and stored the clinical specimens. GC and SC provided the clinical specimens and expertize. XY and XZ provided intellectual discussions. WH conceptualized and designed the project and co-wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Zhang, X., Zhang, L., Du, Y. et al. A novel FOXM1 isoform, FOXM1D, promotes epithelial–mesenchymal transition and metastasis through ROCKs activation in colorectal cancer. Oncogene 36, 807–819 (2017). https://doi.org/10.1038/onc.2016.249
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.249
This article is cited by
-
A patient-derived organoid-based study identified an ASO targeting SNORD14E for endometrial cancer through reducing aberrant FOXM1 Expression and β-catenin nuclear accumulation
Journal of Experimental & Clinical Cancer Research (2023)
-
Targeting the oncogenic transcription factor FOXM1 to improve outcomes in all subtypes of breast cancer
Breast Cancer Research (2023)
-
ERCC6L facilitates the progression of laryngeal squamous cell carcinoma by the binding of FOXM1 and KIF4A
Cell Death Discovery (2023)
-
Peristalsis-Associated Mechanotransduction Drives Malignant Progression of Colorectal Cancer
Cellular and Molecular Bioengineering (2023)
-
Role of Interleukins in Inflammation-Mediated Tumor Immune Microenvironment Modulation in Colorectal Cancer Pathogenesis
Digestive Diseases and Sciences (2023)
