
Abstract
FOXM1 is implicated in genotoxic drug resistance but its mechanism of action remains elusive. We show here that FOXM1-depletion can sensitize breast cancer cells and mouse embryonic fibroblasts (MEFs) into entering epirubicin-induced senescence, with the loss of long-term cell proliferation ability, the accumulation of γH2AX foci, and the induction of senescence-associated β-galactosidase activity and cell morphology. Conversely, reconstitution of FOXM1 in FOXM1-deficient MEFs alleviates the accumulation of senescence-associated γH2AX foci. We also demonstrate that FOXM1 regulates NBS1 at the transcriptional level through an forkhead response element on its promoter. Like FOXM1, NBS1 is overexpressed in the epirubicin-resistant MCF-7EpiR cells and its expression level is low but inducible by epirubicin in MCF-7 cells. Consistently, overexpression of FOXM1 augmented and FOXM1 depletion reduced NBS1 expression and epirubicin-induced ataxia-telangiectasia mutated (ATM)phosphorylation in breast cancer cells. Together these findings suggest that FOXM1 increases NBS1 expression and ATM phosphorylation, possibly through increasing the levels of the MRN(MRE11/RAD50/NBS1) complex. Consistent with this idea, the loss of P-ATM induction by epirubicin in the NBS1-deficient NBS1-LBI fibroblasts can be rescued by NBS1 reconstitution. Resembling FOXM1, NBS1 depletion also rendered MCF-7 and MCF-7EpiR cells more sensitive to epirubicin-induced cellular senescence. In agreement, the DNA repair-defective and senescence phenotypes in FOXM1-deficent cells can be effectively rescued by overexpression of NBS1. Moreover, overexpression of NBS1 and FOXM1 similarly enhanced and their depletion downregulated homologous recombination (HR) DNA repair activity. Crucially, overexpression of FOXM1 failed to augment HR activity in the background of NBS1 depletion, demonstrating that NBS1 is indispensable for the HR function of FOXM1. The physiological relevance of the regulation of NBS1 expression by FOXM1 is further underscored by the strong and significant correlation between nuclear FOXM1 and total NBS1 expression in breast cancer patient samples, further suggesting that NBS1 as a key FOXM1 target gene involved in DNA damage response, genotoxic drug resistance and DNA damage-induced senescence.
Similar content being viewed by others


Introduction
Breast cancer is the most common malignancy among women worldwide, as well as one of the leading causes of cancer death. Genotoxic drugs, including anti-metabolites, topoisomerase inhibitors, and anthracyclines as well as radiotherapy are used universally in the treatment of breast cancer and to reduce the chance of metastasis. These therapeutic agents are particularly important for the treatment of breast cancers that are not suitable for or refractory to endocrine therapies. Anthracyclines, including epirubicin, are anti-cancer antibiotics that have proven efficacy in the treatment of breast, lung and ovarian cancers as well as leukaemia.1 The primary mechanism of action of anthracyclines is to interfere with enzymes involved in DNA replication, but these drugs can also induce DNA intercalation and damage, which will ultimately result in DNA lesions primarily in the forms of double-stranded breaks (DSBs). The cellular response to DNA damage, the so-called ‘DNA damage response’, involves the recognition of DNA damage, the activation of cell cycle checkpoints, transcription programmes, DNA damage repair, and senescence and apoptosis, if the damage is irreparable.2
DSBs are predominantly repaired by two mechanisms, homologous recombination (HR) and non-homologous end joining.3 Ataxia-telangiectasia mutated (ATM) is a phosphatidylinositol 3-kinase-related kinase central to the DNA damage response signalling and is activated upon DNA damage by DSBs. Its activation, accompanied by autophosphorylation on serine 1981 (P-ATM), induces phosphorylation of its downstream target histone H2AX, leading ultimately to the recruitment of DNA repair proteins to the sites of damage.4 In addition, the ATM kinase also directly phosphorylates modulator proteins, including p53BP1, SMC1, BRCA1, NBS1 and CHK2, essential for triggering cell-cycle arrest, apoptosis and DNA repair.5, 6, 7, 8 The MRN (MRE11/RAD50/NBS1) complex has a key role in sensing DSBs and in the activation of ATM.7 The assembly of the MRN complex is a rate-limiting step for the recruitment of the ATM and its phosphorylation, and therefore, the activation of the DNA damage repair response.9 In particular, ATM is dependent on NBS1 as part of the MRN complex for sensing DSBs, and also as an adaptor molecule for phosphorylation of its downstream substrates.10, 11
Cellular senescence is an intrinsic cellular response that restricts unlimited cell proliferation and has a key physiological role in tumour suppression through preventing cancer initiation and progression.12 Cells can be induced to undergo premature senescence by intracellular as well as extracellular stress including irradiation, persistent DNA damage response, oncogene activation, telomere erosion, oxidative stress, toxins and stem cell reprogramming.13 FOXM1 is a member of the Forkhead box (FOX) superfamily of transcription factors that share a conserved winged-helix DNA-binding domain.14 It is ubiquitously expressed in actively proliferating tissues and has a crucial role in a wide range of biological processes, including cell cycle progression, angiogenesis, metastasis, apoptosis, tissue regeneration and drug resistance. We have obtained evidence that FOXM1 can protect cells from genotoxic agent-induced senescence by enhancing HR DNA repair. Consistently, FOXM1 is overexpressed in genotoxic agent-resistant cancer cells. In here, we studied the role of FOXM1 in genotoxic drug resistance and found that FOXM1 regulates NBS1 expression to modulate HR DNA damage repair, which has a role in overcoming DNA damage agent-induced cellular senescence.
Results
FOXM1 deletion decreases long-term viability and activates cellular senescence in response to DNA damage
Recent evidence shows that FOXM1 can protect cells from genotoxic agent-induced cell death by enhancing DNA damage repair.15, 16, 17 However, the role of FOXM1 in promoting long-term cell viability in response to genotoxic treatment has not been evaluated. To address this, we treated wild-type (WT) and Foxm1−/− MEFs with a range of concentrations of epirubicin and studied their long-term viability by clonogenic assays (Supplementary Figure S1). The results showed that at 0, 20 and 40 nM epirubicin, the colony formation capacity of Foxm1−/− MEFs was significantly impaired compared with WT MEFs (Figure 1a). To confirm this further, WT and Foxm1−/− MEFs were also exposed to moderate levels of ionizing radiation (Figure 1b). At 5 Gy γ-irradiation, the colony formation capacity of Foxm1−/− MEFs was significantly reduced compared with WT MEFs. As these low epirubicin concentrations and ionizing irradiation doses do not induce considerable cell death,15 we reasoned that epirubicin might induce long-term proliferative arrest, implicative of cellular senescence. To test this conjecture, WT and Foxm1−/− MEFs were assayed for senescence-associated β-galactosidase (SAβ-gal) activity following low doses of epirubicin treatment (Figure 1c). At 20 and 40 nM epirubicin treatment, significantly higher numbers of Foxm1−/− MEFs displayed distinct SAβ-gal activity and ‘flat cell’ morphology compared with WT MEFs, suggesting that FOXM1 has a key role in DNA damage-induced senescence. Moreover, significantly higher numbers of Foxm1−/− MEFs displayed SAβ-gal and senescent morphology compared with WT MEFs following exposure to 5 Gy of γ-irradiation (Figure 1d). Senescent cells accumulate γH2AX foci as markers for unrepaired or irreparable damaged DNA.18 We also found that Foxm1−/− MEFs accumulated significantly higher numbers of γH2AX foci compared with WT MEFs at prolonged times of 24, 48 and 72 h following epirubicin treatment, suggesting that FOXM1 deletion renders MEFs more susceptible to the accumulation of DSBs induced by genotoxic agents (Supplementary Figure S2).

FOXM1 deletion inhibits colony formation and induces cellular senescence in response to DNA damage in MEFs. (a) Clonogenic assays were performed to assess the colony formation efficiency of Foxm1−/− and WT MEFs. A total of 2000 cells were seeded in six-well plates, treated with 0, 20 and 40 nM of epirubicin and grown for 15 days. The cells were then stained with crystal violet (left panel). The result (right panel) represents average of three independent experiments±s.d. Statistical significance was determined by Student’s t-test (**P⩽0.01). (b) Clonogenic assay of Foxm1−/− and WT MEFs were either non-radiated or exposed to 5 Gy of γ-irradiation. (c) SAβ-gal staining of Foxm1−/− and WT MEFs treated with 0, 20 and 40 nM of epirubicin. 15 days after treatment, cells were stained for SAβ-gal activities. (d) SAβ-gal staining of Foxm1−/− and WT MEFs treated with γ-irradiation (0 and 5 Gy). The graphs c and d show the percentage of SAβ-gal-positive cells as measured from five different fields from two independent experiments. Bars represent average±s.d. Statistical significance was determined by Student’s t-test (**P⩽0.01, significant; n.s., non-significant).
FOXM1 depletion resensitizes MCF-7 breast cancer cells to epirubicin-induced cellular senescence
We next studied the effects of FOXM1 depletion by small interfering RNA (siRNA) on the long-term viability of MCF-7 and the epirubicin-resistant MCF-7EpiR breast cancer cell lines by clonogenic assays (Figure 2). FOXM1-knockdown sensitized MCF-7 cells to long-term proliferative arrest at relatively low epirubicin concentrations (for example, 10 and 20 nM; Figure 2a), whereas at higher concentrations (>20 nM) the colony formation ability was completely lost in MCF-7 cells (data not shown). FOXM1 depletion alone almost completely abolished the colony formation ability of MCF-7EpiR cells irrespective of the dosage of epirubicin used, suggesting that MCF-7EpiR cells are dependent on high FOXM1 expression for long-term survival (Figure 2b). In addition, similar to what it occur in MEFs, FOXM1 knockdown in MCF-7 cells enhanced the number of cells exhibiting SAβ-gal activity and morphology at 0 and 10 nM epirubicin (Figure 2c). Consistent with previous results, FOXM1 knockdown in MCF-7EpiR cells resulted in almost all cells displaying senescence-associated SAβ-gal activity and morphology independent of epirubicin levels, suggesting MCF-7EpiR cells have become dependent on FOXM1 to override senescence (Figure 2d). Senescent cells accumulate γH2AX foci as markers for unrepaired or irreparable damaged DNA.18 To confirm this further, we studied the effects of FOXM1 depletion on nuclear γH2AX foci formation in MCF-7 and MCF-7EpiR cells following γ-irradiation (Figures 3a and b). Quantification of γH2AX foci showed that γ-irradiation induced the accumulation of significantly more foci over the extended time course of 24, 48 and 72 h in FOXM1-knockdown cells in comparison to control siRNA-transfected MCF-7 and MCF-7EpiR cells, further indicating that FOXM1 has a critical role in overcoming DNA damage-induced senescence.

Knockdown of FOXM1 suppresses growth and induces senescence in MCF-7 and MCF-7EpiR cells. (a) MCF-7 and (b) MCF-7EpiR were transfected with NS (non-targeting) siRNA or FOXM1 siRNA. Twenty-four hours after transfection, cells were seeded in six-well plates, treated with epirubicin, grown for 15 days and then stained with crystal violet (left panel). The results (right panel) represent average of three independent experiments±s.d. Statistical significance was determined by Student’s t-test (*P⩽0.05, **P⩽0.01, ***P⩽0.005; n.s., non-significant). In parallel, (c) MCF-7 and (d) MCF-7EpiR transfected with NS siRNA or siFOXM1 were seeded in six-well plates, treated with epirubicin. Five days after treatment, cells were stained for SAβ-gal activity. The graphs c and d show the percentage of SAβ-gal-positive cells as measured from five different fields from two independent experiments. Bars represent average±s.d. Statistical significance was determined by Student’s t-test (*P⩽0.05, **P⩽0.01, ***P⩽0.005, significant; n.s., non-significant).

FOXM1 depletion in MCF-7 cells causes the accumulation of significantly higher numbers of γH2AX foci in response to γ-irradiation. (a) MCF-7 and (b) MCF-7EpiR were transfected with NS siRNA or FOXM1 siRNA. Twenty-four hours after transfection, cells cultured on chamber slides were either non-radiated or exposed to 5 Gy of γ-irradiation for 24, 48 and 72 h. Cells were then fixed and immunostained for γH2AX foci (green). Nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI; blue). Images were acquired with Leica TCS SP5 ( × 63 magnification). For each time point, images of at least 100 cells were captured and used for quantification of γH2AX foci number. Results represent average of three independent experiments±s.d. Statistical analyses were conducted using Student’s t-tests against the correspondent time point (**P⩽0.01, significant; n.s., non-significant).
FOXM1 enhances NBS1 and P-ATM expression to promote DNA damage repair signalling
We have shown previously that FOXM1 is essential for HR repair after DSBs.17 To elucidate the underlying mechanistic role of FOXM1 in DNA damage-induced senescence and epirubicin resistance, we next compared the expression patterns of FOXM1 with those of the early HR sensor proteins, including ATM and components of the MRN (MRE11/RAD50/NBS1; NBS1 also known as Nibrin and NBN) complex in MCF-7 and MCF-7EpiR cells in response to epirubicin. Western blot analysis showed that the expression level of FOXM1 protein was induced transiently before declining eventually following epirubicin treatment in MCF-7 cells, whereas FOXM1 was expressed consistently at high levels in the resistant MCF-7EpiR cells (Figure 4a). ATM was expressed at higher levels in MCF-7EpiR compared with the control MCF-7 cells. Autophosphorylated ATM (P-ATM), indicative of ATM activity, was constitutively high in MCF-7EpiR cells, whereas the P-ATM signal was low but inducible by epirubicin in MCF-7 cells. NBS1 as well as its active phosphorylated form P-NBS1 followed similar kinetics as FOXM1 upon epirubicin treatment in both MCF-7 and MCF-7EpiR cells (Figure 4a; Supplementary Figure S3), suggesting the regulation of NBS1 expression and activity by FOXM1. By contrast, the other MRN components, MRE11 and RAD50, did not show substantial correlation with the expression patterns of FOXM1 upon epirubicin treatment. Consistently, quantitative real-time PCR (qRT–PCR) analysis revealed that upon epirubicin treatment the NBS1 and FOXM1 mRNA displayed similar kinetics in both MCF-7 and MCF-7EpiR cells, further suggesting FOXM1 may transcriptionally regulate NBS1 expression (Figure 4b). To test this idea, both MCF-7 and MCF-7EpiR cells were transfected with non-targeting control (non-specific (NS)) or FOXM1 siRNA pools for 24 h and were subjects to western blot and qRT–PCR analyses after incubation with epirubicin or vehicle control for another 24 h. Silencing the expression of FOXM1 caused a significant reduction in the NBS1 protein and mRNA levels in both MCF-7 and MCF-7EpiR cells, whereas only a moderate decrease was observed for the RAD50 and MRE11 protein, but not at the mRNA level, upon FOXM1 knockdown (Figures 4c and d; Supplementary Figure S4). This suggests that in contrast to NBS1, RAD50 and MRE11 are not directly regulated by FOXM1, raising the possibility that their protein expression levels might be stabilized through forming an active MRN complex. Notably, the expression of P-ATM and P-CHK2 was induced in MCF-7 and MCF-7EpiR cells after epirubicin treatment. This is likely to reflect the genotoxic stress response of the cells after epirubicin treatment following the serum starvation conditions used for siRNA transfection. These stress conditions might also be responsible for the high levels of cell death and global protein degradation observed in the MCF-7 cells. It is also noteworthy that P-ATM levels were induced by epirubicin treatment, but this induction was abolished upon FOXM1 depletion by siRNA, suggesting FOXM1 is involved in the activation of ATM. Similar results were also observed for the TERT48 human fibroblasts (Supplementary Figure S5). Furthermore, the NBS1 mRNA was also downregulated after FOXM1 depletion in the breast cancer cell lines, MDA-MB-231 and ZR-75-1 cells (Supplementary Figure S5). To confirm further that FOXM1 modulates ATM activity through controlling NBS1 expression, we examined the expression of these early DNA damage sensor proteins in WT and Foxm1−/− MEFs following epirubicin treatment (Figure 5a; Supplementary Figure S6). Consistent with results from the breast cancer cell lines, NBS1 and P-NBS1 accumulated with similar kinetics as FOXM1 in WT MEFs following epirubicin treatment, but were barely detectable in Foxm1−/− MEFs. By contrast, RAD50 and MRE11 were expressed at only marginally lower levels in Foxm1−/− MEFs compared with WT MEFs. The western blot results also showed that although ATM expressed at comparable levels in WT and Foxm1−/− MEFs, P-ATM was induced by epirubicin and expressed at much higher levels in WT compared with Foxm1−/− MEFs. qRT–PCR analysis also indicated that upon epirubicin treatment the NBS1 and FOXM1 mRNA displayed similar kinetics in both WT and Foxm1−/− MEFs, further confirming FOXM1 regulates NBS1 expression (Figure 5b). Notably, the slight discrepancies between FOXM1 and NBS1 protein and RNA levels can be due to the fact that both FOXM1 and NBS1 expression as well as activity are also regulated by post-transcriptional mechanisms.17, 19, 20 Nevertheless, together these findings led us to postulate that FOXM1 may target NBS1 expression at the transcriptional level to enhance DNA damage repair signalling and genotoxic drug resistance.

FOXM1 regulates NBS1 expression and ATM activity in MCF-7 and MCF7-EpiR cells. (a) MCF-7 and MCF7-EpiR cells were treated with 1 μM epirubicin for 0, 4, 8, 16, 24 and 48 h. Following treatment, the expression levels of FOXM1, NBS1, P-NBS1, RAD50, MRE11, P-ATM, ATM and β-tubulin were determined by western blotting. (b) Real-time quantitative PCR (qRT–PCR) was used to determine FOXM1 and NBS1 mRNA transcript levels (normalized against L19 mRNA levels). Bars represent the mean+s.d. of three independent experiments. (c, d) MCF-7 and MCF7EpiR cells were transfected with either NS siRNA or FOXM1 siRNA. Twenty-four hours after transfection, the cells were either untreated or treated with 1 μM epirubicin for 24 h. (c) The protein expression was determined by western blot analysis using antibodies against FOXM1, NBS1, P-NBS1, RAD50, MRE11, P-ATM, ATM, P-CHK2,CHK2 and β-tubulin. (d) FOXM1, NBS1, MRE11 and RAD50 transcripts were next analysed by qRT–PCR in the untreated cells (all gene transcripts were normalized against L19). Statistical significance was determined by Student’s t-test (**P⩽0.01, ***P⩽0.005, significant; n.s., non-significant).

NBS1 and P-ATM expression are downregulated in FOXM1-deficient MEFs. (a) WT and Foxm1−/− MEFs were treated with 0.1 μM epirubicin for 0, 0.5, 1, 2, 4, 16 and 24 h. The harvested cells were subjected to western blot analysis, and the protein expression levels of FOXM1, NBS1, P-NBS1, RAD50, MRE11, P-ATM, ATM and β-tubulin were determined. (b) WT and Foxm1−/− MEFs were exposed to 0.1 μM of epirubicin for 0, 1, 2, 4, 8, 24 and 48 h and the mRNA transcript levels of FOXM1 and NBS1 determined by qRT–PCR after normalizing against L19 the house keeping gene. Bars represent average±s.d. Statistical significance was determined by Student’s t-test.
FOXM1 modulates NBS1 expression and ATM phosphorylation
We next overexpressed FOXM1 in the MCF-7 cells and examined its effects on the NBS1 expression and ATM activity. The result showed that overexpression of FOXM1 caused an induction of NBS1 levels and ATM phosphorylation in MCF-7 cells (Figure 6a; Supplementary Figure S7), further confirming that FOXM1 regulates NBS1 expression and thus MRN complex formation to promote ATM activation and phosphorylation. It is notable that despite FOXM1 overexpression, NBS1 levels decreased after 48 h of epirubicin treatment, probably because both FOXM1 and NBS1 are also regulated at post-transcriptional levels in response to epirubicin.19 We next studied the effects of NBS1 depletion in the resistant MCF-7EpiR cells (Figure 6b). Knockdown of NBS1 expression by siRNA did not affect FOXM1, RAD50 and MRE11 expression but resulted in a downregulation of P-ATM and P-NBS expression. We also showed that the NBS1 is required for ATM activition in response to epirubicin by demonstrating that reconstituting NBS1 expression can restore ATM activity and phosphorylation in response to epirubicin in the NBS1-deficient NBS1-LBI cells (Supplementary Figure S8).

FOXM1 regulates ATM phosphorylation and NBS1 expression through a FHRE site within its promoter. (a) MCF-7 cells were transfected with pcDNA3 empty vector or pcDNA3-FOXM1 plasmids following which the cells were subjected to 1 μM epirubicin treatment for 0, 6 24 and 48 h. Western blot analyses were performed to analyse the protein expression level changes of FOXM1, NBS1, P-ATM, ATM, RAD50, MRE11 and β-tubulin. (b) MCF7-EpiR cells were transfected with NS siRNA or NBS1 siRNA. Twenty-four hours after the transfection, cells were treated with 1 μM epirubicin for 0, 4, 24 and 48 h and harvested for western blot analyses. Protein expression levels of the indicated proteins: NBS1, P-NBS1, FOXM1, ATM, P-ATM, RAD50, MRE11, PARP and β-tubulin were analysed (arrow indicates cleaved PARP proteins). (c) MCF-7 cells were transciently transfected with 20 ng of pGL3-NBS1 WT or pGL3-NBS1 mut promoter together with the control Renilla plasmid and increasing amounts (0, 10, 15 and 20 ng) of FOXM1 expression vector. After 24 h, cells were assayed for luciferase activity. The relative luciferase activity was calculated after normalizing with the control Renilla activity. (d) MCF-7 either untransfected or transfected with pcDNA3-FOXM1, and MCF7-EpiR either untreated or treated with 1 μM epirubicin for 16 h were used for chromatin immunoprecipitation assays using the IgG as negative control and anti-FOXM1 antibody, as indicated. After reversal of cross-linking, the coimmunoprecipitated DNA was amplified by PCR, using primers amplifying the FOXM1 FHRE-binding site containing region (−300/−24 bp) and a control region (−1097/−826 pb), and resolved in 2% agarose gel. Inverted ethidium bromide stained images are shown.
FOXM1 regulates NBS1 expression through a forkhead response element (FHRE) in its promoter
We next investigated if the regulation of NBS1 by FOXM1 is at the promoter level. To this end, MCF-7 cells were transiently co-transfected with the FOXM1 expression construct and a luciferase reporter gene under the control of either a human NBS1 WT or a mutant (mut) NBS1 (1.5 kbp) promoter with a putative FHRE (−78 bp) mutated (Figure 6c).21 We observed the (WT) NBS1 promoter activity was augmented by FOXM1 in a dose-dependent manner, whereas the mut NBS1 promoter had lower basal promoter activity and was not inducible by FOXM1 (Figure 6c). Collectively, these results suggest that FOXM1 is able to transactivate NBS1 gene through the FHRE located at position −78 bp, providing evidence that NBS1 is a direct target gene of FOXM1. To confirm further that FOXM1 binds to the FHRE of the endogenous NBS1 promoter in vivo, we studied the occupancy of the FHRE region of the NBS1 promoter by FOXM1 using chromatin immunoprecipitation with and without FOXM1 transfection in the MCF-7 cells and before and after 16 h epirubicin treatment in the MCF-7EpiR cells. The chromatin immunoprecipitation analysis showed that FOXM1 is recruited to the endogenous FHRE in both the MCF-7 and MCF-7EpiR and its binding to the FHRE increases in response to epirubicin (Figure 6d). Together, these findings indicate that NBS1 is a direct in vivo transcriptional target of FOXM1.
Correlation between NBS1 and FOXM1 expression in breast cancer samples
After establishing that NBS1 is a direct transcriptional target of FOXM1 in breast cancer and fibroblast cell lines, the correlation of FOXM1 and NBS1 expression were assessed by immunohistochemistry in breast cancer samples from 116 patients (Figure 7a).22 FOXM1 and NBS1 staining were detected in both nuclear and cytoplasmic compartments. Statistical analysis of the expression patterns revealed that there was a strong and significant correlation between FOXM1 nuclear staining and total NBS1 staining ((Pearson coefficient=0.318, P=0.002), providing further physiological evidence that FOXM1 regulates NBS1 expression in breast cancer patient samples (Figure 7a)). In addition, there was also a trend linking high NBS1 nuclear localization expression to poor survival (P=0.164 for overall survival, log-rank test, Kaplan–Meier estimate analysis), which by multivariate Cox regression analysis, when adjusted by patients’ T-stage and lymph-node involvement was significantly correlated with poorer survival (relative risk (RR)=2.869, P=0.048; Supplementary Figure S9). Further analysis of FOXM1 and NBS1 mRNA transcript expression in another previously published cohort (2878 breast cancer patients)23 revealed that high FOXM1 and NBS1 mRNA expression levels are very significantly associated with poor survival (P<0.0001 and P=0.0012, respectively, for overall survival, Kaplan–Meier analysis; Supplementary Figure S10). The significance of NBS1 in survival analyses suggests direct involvement of NBS1 in regulating cell senescence and DNA damage repair in genotoxic drug response.

Correlation between NBS1 and FOXM1 expression in breast cancer samples. (a) Correlation of FOXM1 and NBS1 expression were assessed and scored by immunohistochemistry in breast cancer samples from 116 patients. FOXM1 and NBS1 staining were detected in both nuclear and cytoplasmic compartments. Statistical analysis of the expression patterns revealed that there was a strong and significant correlation between FOXM1 nuclear staining and total NBS1 staining (Pearson coefficient=0.318, P=0.002). (b) NBS1 depletion induces senescence-associated phenotypes in MCF-7 breast cancer cells. MCF-7 and MCF-7 EpiR were transfected with NS siRNA or NBS1 siRNA. 24 h after transfection, 2000 cells were seeded in six-well plates, treated with epirubicin, grown for 15 days and then stained with crystal violet (left panel). The result (right panel) represents average of three independent experiments±s.d. Statistical significance was determined by Student’s t-test (**P⩽0.01, significant; n.s., non-significant). In parallel, (c) MCF-7 and MCF-7 EpiR transfected with NS siRNA or siNBS1 were seeded in six-well plates, treated with epirubicin for 5 days. Cells were stained for SAβ-gal activities. The graphs show the percentage of SAβ-gal-positive cells as measured from five different fields from two independent experiments. Bars represent average±s.d. Statistical significance was determined by Student’s t-test (***P⩽0.005, significant).
NBS1 depletion induces senescence-associated phenotypes in MCF-7 breast cancer cells
Given that FOXM1 is involved in DNA damage-induced senescence after long treatments with low levels of genotoxic agents and also controls NBS1 expression, we next explore the role of NBS1 in genotoxic drug-induced senescence, we next studied the effects of NBS1 depletion on the long-term viability of MCF-7 and MCF-7EpiR cells by clonogenic assays (Figure 7b). Like FOXM1, NBS1-knockdown sensitized MCF-7 and MCF-7EpiR cells to long-term proliferative arrest following treatment with epirubicin (Figure 7b). Consistently, NBS1 knockdown in MCF-7 and MCF-7EpiR cells also enhanced the number of cells exhibiting SAβ-gal activity and morphology at 0 and 10 nM epirubicin (Figure 7c). These findings strongly suggest that NBS1 protects MCF-7 and MCF-7EpiR cells from entering senescence.
NBS1 and FOXM1 are required for HR repair
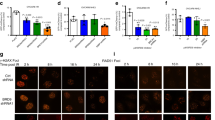
We have shown previously that FOXM1 is essential for HR but not non-homologous end joining DNA repair.17 We next analysed the role of NBS1 and FOXM1 in DSB repair using HeLa cell lines harbouring an integrated direct repeat green fluorescent protein (DR-GFP) reporter for HR.24, 25 The I-SceI expression plasmid was cotransfected into DR-GFP HeLa cells with the NS, FOXM1 or NBS1 siRNA and the percentage of GFP-positive cells assayed by flow cytometry (Supplementary Figure S11). Knockdown of NBS1 or FOXM1 using siRNA significantly decreased the number of GFP-positive cells in comparison with NS control siRNA in the HR DR-GFP reporter system (54.0 and 67.6%, respectively; Figure 8a). Conversely, overexpression of NBS1 and FOXM1 significantly increased the DSB repair via HR by 82.9% and 87.3%, respectively (Figure 8a). These observations indicate that similar to FOXM1, NBS1 contributes to HR-directed DSB repair. To investigate the functional relationship between NBS1 and FOXM1 in HR-directed DSB repair, we next examined the effects of NBS1 depletion in the background of FOXM1 overexpression using DR-GFP HeLa reporter system. The results showed that the HR activity after NBS1 knockdown and FOXM1 overexpression (49.4%) was not significantly higher than NBS1 depletion alone, indicating that the function of FOXM1 to direct HR repair is linked to NBS1.

NBS1 and FOXM1 are required for homologous recombination (HR) repair. (a) HR repair was assayed in HeLa cells harbouring a DR-GFP reporter system. Cells were either transfected with control pcDNA3, pcDNA3-FOXM1 or pFlag-NBS1 or with NS siRNA, FOXM1 siRNA or NBS1 siRNA, or with co-transfection control, pFlag-NBS1 plus FOXM1 siRNA. 48 h after transfection, the cells were transfected with I-Scel plasmid. Cleavage of an I-Scel and repair by HR leads to GFP expression in cells. The percentage of GFP-positive cells was determined by fluorescence-activated cell sorting analysis at day 3 post-transection. Bars are average±s.d. of three independent experiments. In parallel, cells were also harvested for western blot analysis. (b) Ectopic expression of NBS1 in Foxm1−/− MEFs reduces the accumulation of γH2AX foci. Foxm1−/− MEFs were either transfected with pmCherry control plasmids or co-transfected with pmCherry and NBS1 plasmids (Red) and treated with 0.1 μM of epirubicin for 0, 4, 24, 48 and 72 h. The cells were then immunostained for γH2AX foci (Green) and nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI; blue) to determine DNA double-strand breaks. γH2AX foci quantification is shown. Bars represent the average of γH2AX foci per cell from three independent experiments±s.d. Statistical significance was determined by Student’s t-test (*P⩽0.05, **P⩽0.01, ***P⩽0.005, significant; n.s., non-significant).
Ectopic expression of NBS1 in Foxm1−/− MEFs abrogates the accumulation of γH2AX foci
Finally, to demonstrate that the downregulation of NBS1 expression in FOXM1-depleted cells is responsible for the accumulation of senescence-associated γH2AX foci upon epirubicin treatment, we next determined whether overexpression of NBS1 in Foxm1−/− MEFs is able to alleviate the accumulation of γH2AX foci particularly at the longer times of 24, 48 and 72 h (Figure 8b). Strikingly, Foxm1−/− MEFs co-transfected with pmCherry (red) and NBS1 displayed significantly fewer foci compared with the adjacent/neighbouring non-transfected cells at the longer time points. The overexpression of NBS1 had the same effects as reconstituting Foxm1−/− MEFs with pmCherry-FOXM1 (Supplementary Figure S12). In contrast, Foxm1−/− MEFs transfected with the empty-pmCherry control have similar kinetics for senescence-associated γH2AX foci accumulation as the non-transfected cells (Supplementary Figure S12). Taken together, these findings further suggest that the role of FOXM1 to repair DSB-DNA damage is related to its ability to control NBS1 expression.
Discussion
Senescence is an intrinsic cellular response that induces irreversible cell-cycle arrest. Cellular senescence restricts unlimited cell proliferation and has a critical role in both ageing as well as tumour suppression. It was first observed in cell culture, but has also been proved to exist in vivo. It is generally perceived that most cancer as well as tissue culture cells have overcome cellular senescence and achieved immortality.12 However, the present study shows that breast cancer cell lines as well as immortalized MEFs can be induced into entering cellular senescence by DNA-damaging agents, including epirubicin and ionizing irradiation. Notably, the doses of DNA-damaging agents required for triggering senescent phenotypes in these cells are substantially lower than that required to induce short-term proliferative arrest and cell death,15, 17 but are in line with the physiological doses used in the clinic,26 raising the possibility that cellular senescence may be the predominant mechanism of action for the genotoxic anti-cancer agents.
FOXM1 has been implicated in genotoxic drug resistance but its role and mechanism of action are still not yet fully understood. We show here that FOXM1 has a central role in modulating DNA damage-induced senescence and thus genotoxic agent resistance. Depletion of FOXM1 sensitized MCF-7 breast cancer cells as well as MEFs into entering epirubicin-induced senescence, as revealed by the loss in long-term cell proliferation ability, the time-dependent accumulation of γH2AX foci, and induction of SAβ-gal activity and cell morphology. Conversely, reconstitution of FOXM1 in FOXM1-deficient MEFs alleviated the accumulation of senescence-associated γH2AX foci. NBS1 is a component of the MRE11/RAD50/NBS1 complex, which has a central role in processing damaged DNA for repair and in promoting ATM-dependent DNA damage response signalling.27 In defining FOXM1 targets that have a role in genotoxic agent-induced senescence, we identified NBS1 as a key FOXM1 target involved in DNA damage repair, genotoxic drug resistance and DNA damage-induced senescence. At the molecular level, we found that FOXM1 regulates NBS1 at the protein and mRNA levels through an FHRE on its promoter in response to epirubicin treatment. Consistently, overexpression of FOXM1 augmented NBS1 expression and ATM phosphorylation, whereas FOXM1 depletion reduced NBS1 expression and ATM phosphorylation upon epirubicin treatment in breast cancer cells. Like FOXM1, NBS1 was overexpressed in the drug-resistant MCF-7EpiR cells and its expression level was low but inducible by epirubicin in MCF-7 cells. We also found that NBS1, but not MRE11 and RAD50, levels were modulated by FOXM1 and in response to epirubicin, suggesting that NBS1 is limiting for the formation of the MRN complex and that its level sets the threshold for DNA repair activity. Collectively, these findings suggest that FOXM1 increases NBS1 expression and ATM phosphorylation, possibly mediated by increasing the levels of the MRN complex. Consistent with this idea, the loss of P-ATM induction upon epirubicin treatment in the NBS1-deficient NBS1-LBI human fibroblasts can be rescued by NBS1 reconstitution.
Resembling FOXM1, NBS1 depletion also rendered MCF-7 and MCF-7EpiR cells more sensitive to epirubicin-induced cellular senescence. In agreement, the DNA repair defect and epirubicin-induced senescence phenotypes in FOXM1-deficent cells could be effectively rescued by overexpression of NBS1. Consistently, overexpression of NBS1 and FOXM1 similarly enhanced and their depletion downregulated HR repair. Importantly, overexpression of FOXM1 failed to augment HR activity in the background of NBS1 depletion, demonstrating that NBS1 is indispensable for the HR function of FOXM1. Consistent with our findings, a role in DNA damage-induced senescence-associated inflammatory cytokine secretion has previously been suggested for NBS1.28 The physiological relevance of the regulation of NBS1 expression by FOXM1 is further underscored by the strong and significant correlation between nuclear FOXM1 and total NBS1 expression in breast cancer patient samples. Furthermore, both FOXM1 and NBS1 mRNA expression are significantly associated with poor prognosis in breast cancer, supporting a physiological role of FOXM1 and its target NBS1 in genotoxic drug resistance. In summary, we identify NBS1 as a key FOXM1 target gene involved in DNA damage response, genotoxic drug resistance and DNA damage-induced senescence. Our data also suggest that the FOXM1-NBS1 axis can be a reliable prognostic marker and a viable target for therapeutic intervention for targeting cancer and for overcoming genotoxic agent resistance in cancer.
Materials and Methods
Cell culture
The human breast carcinoma MCF-7, MDA-MB-231 and ZR-75-1 cell lines were originated from the American Type Culture Collection (Manassas, VA, USA) and were acquired from the Cell Culture Service, Cancer Research UK, where it was tested and authenticated. MEFs isolated from Foxm1−/− and WT mice have been previously described.29 The 48BRhtert and NBS1-LBI human fibroblasts have also been described.30 The HeLa DR-GFP reporter cells were a gift from Dr Maria Jasin (Memorial Sloan-Kettering Cancer Center, New York, USA).24, 31 All cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin/streptomycin and maintained at 37 °C in a humidified incubator with 10% CO2. The MCF-7EpiR was maintained in 10 μmol/l Epirubicin (Medac, Hamburg, Germany), as previously described.32
Plasmids
The pFlag-Nbs1 and the pcDNA3-FOXM1 have been described.16, 33 The pmCherry-FOXM1 was generated by cloning the full-length FOXM1 cDNA from pcDNA3-FOXM1 into the EcoRI and BamHI sites of the pmCherry-N1 vector (Clontech, Mountain View, CA, USA) and the pCMV-I-SceI has been described.24, 31 The WT NBS1 promoter luciferase reporter construct, pNBSLuc1500, has previously been described.21 Expression plasmid transfections were performed with FuGENE 6 (Roche, Indianapolis, IN, USA) according to the manufacturer’s recommendations. See also Supplementary Materials and Methods for mut NBS1 promoter generation.
Luciferase reporter assay
Cells were co-transfected with the human NBS1 promoter luciferase reporter and transfection control Renilla (pRL-TK; Promega, Southampton, UK) constructs using FuGENE6 (Roche). For promoter analysis, 24 h after transfection, cells were then collected, washed twice in PBS and harvested for firefly/Renilla luciferase assays using the Dual-Glo Luciferase reporter assay system (Promega, Madison, WI, USA) according to manufacturer’s instruction. Luminescence was then read using the 9904 TOPCOUNT Perkin Elmer (Beaconsfield, UK) plate reader.
γH2AX immunofluorescent staining and foci quantification
Cells grown on chamber culture slides were fixed in 4% paraformaldehyde (Thermo Scientific, Rockford, IL, USA) for 15 min followed by permeabilization for 10 min with 0.2% Triton X-100 in PBS, and blocking with 5% goat serum for 30 min at room temperature. The slides were incubated with primary antibody anti-γH2AX Ser139 (20E3; Cell Signaling, Danvers, MA, USA) overnight at 4 °C. See also Supplementary Materials and Methods.
SAβ-gal assay
Cells were seeded in six-well plates at a density of approximately 20 000 cells/well before treatment with epirubicin or γ-irradiation for 48 h. After culture for a further 5 days, cells were fixed and stained using a Senescence β-Galactosidase Staining Kit #9860 purchased from Cell Signalling Technology (Beverley, MA, USA). Plates were incubated overnight at 37 °C in a dry incubator (no CO2). Cells were then detected for blue staining under a bright-field microscope. The percentage of SAβ-gal-positive cells was calculated by counting the cells in five random fields.
Western blotting and antibodies
Western blotting was performed on whole-cell extracts by lysing cells in buffer as described.34 Antibodies FOXM1 (C-20), β-tubulin (H-235) and I-SceI were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for P-ATM (Ser1981; MAB3806), P-H2AX (Ser139; JBW301) were obtained from Upstate (Millipore, Oxford, UK). The NBS1 (3002) and P-NBS1 antibodies were purchased from Cell Signaling Technology (New England Biolabs Ltd. Hitchin, UK). Total ATM antibody was obtained from Calbiochem (Millipore, Oxford, UK), and MRE11 and RAD50 were from Novus Biologicals (Cambridge, UK). Primary antibodies were detected using horseradish peroxidase-linked anti-mouse or anti-rabbit conjugates as appropriate (Dako, Glostrup, Denmark) and visualized using the ECL detection system (Amersham Biosciences, Pollards Wood, UK).
Quantitative real-time PCR (qRT–PCR)
Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Hilden, Germany). Complementary DNA generated by Superscript III reverse transcriptase and oligo-dT primers (Invitrogen, Paisley, UK) was analysed by qRT–PCR as described.35 Transcript levels were quantified using the standard curve method. For PCR-primer sequences, see Supplementary Materials and Methods.
Gene silencing with siRNAs
For gene silencing, cells were transiently transfected with siRNA SMARTpool reagents purchased from Thermo Scientific Dharmacon (Lafayette, CO, USA) using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. SMARTPool siRNAs used were: siRNA FOXM1 (L-009762-00), siRNA NBS1 (L-009641-00) and the NS control siRNA, confirmed to have minimal targeting of known genes (D-001810-10-05).
Two-step cross-linking chromatin immunoprecipitation
Dual cross-linking chromatin immunoprecipitation using formaldehyde and di-(N-succimidyl) glutarate was performed on MCF-7 and MCF-7EpiR cells, as described.36 See also Supplementary Materials and Methods.
Clonogenic assay
See Supplementary Materials and Methods.
Statistical analysis
Statistical analysis was performed using the SPSS programme version 17. The correlation between FOXM1 and NBS1 expression was analysed by bi-variate Pearson Correlation analysis. Survival analysis was assessed by Kaplan–Meier analysis using log-rank test. P values of less than or equal to 0.05 were considered statistically significant. The statistical significance of differences between the means of two groups was evaluated by paired Student’s t-test using the Microsoft Excel programme and considered significant when *P⩽0.05s, **P⩽0.01 and ***P⩽0.005. n.s. for non-significant.
HR repair assays
HR assays were performed as previously described.24, 31 Analysis was conducted using a FACSCanto analyzer. GFP-fluorescence was analysed using Cell Diva software (Becton Dickinson, Oxford, UK).
Tissue microarray, immnohistochemistry and staining scoring
References
Stearns V, Davidson NE, Flockhart DA . Pharmacogenetics in the treatment of breast cancer. Pharmacogenomics J 2004; 4: 143–153.
Seviour EG, Lin SY . The DNA damage response: balancing the scale between cancer and ageing. Aging (Albany NY) 2010; 2: 900–907.
Helleday T, Lo J, van Gent DC, Engelward BP . DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007; 6: 923–935.
So S, Davis AJ, Chen DJ . Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol 2009; 187: 977–990.
Riches LC, Lynch AM, Gooderham NJ . Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 2008; 23: 331–339.
Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB . Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev 2004; 18: 1423–1438.
Lee JH, Paull TT . ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308: 551–554.
Matsuoka S, Huang M, Elledge SJ . Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282: 1893–1897.
Dupre A, Boyer-Chatenet L, Gautier J . Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat Struct Mol Biol 2006; 13: 451–457.
Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998; 93: 477–486.
Kobayashi J, Antoccia A, Tauchi H, Matsuura S, Komatsu K . NBS1 and its functional role in the DNA damage response. DNA Repair (Amst) 2004; 3: 855–861.
Collado M, Serrano M . Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10: 51–57.
Rodier F, Campisi J . Four faces of cellular senescence. J Cell Biol 2011; 192: 547–556.
Lam EW, Brosens JJ, Gomes AR, Koo CY . Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer 2013; 13: 482–495.
Millour J, de Olano N, Horimoto Y, Monteiro LJ, Langer JK, Aligue R et al. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol Cancer Ther 2011; 10: 1046–1058.
Kwok JM, Peck B, Monteiro LJ, Schwenen HD, Millour J, Coombes RC et al. FOXM1 confers acquired cisplatin resistance in breast cancer cells. Mol Cancer Res 2010; 8: 24–34.
Monteiro LJ, Khongkow P, Kongsema M, Morris JR, Man C, Weekes D et al. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene 2012; 32: 4634–4645.
Kuo LJ, Yang LX . Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 2008; 22: 305–309.
de Olano N, Koo CY, Monteiro LJ, Pinto PH, Gomes AR, Aligue R et al. The p38 MAPK-MK2 axis regulates E2F1 and FOXM1 expression after epirubicin treatment. Mol Cancer Res 2012; 10: 1189–1202.
Gorospe M, de Cabot R . AsSIRTing the DNA damage response. Trends Cell Biol 2008; 18: 77–83.
Chiang YC, Teng SC, Su YN, Hsieh FJ, Wu KJ . c-Myc directly regulates the transcription of the NBS1 gene involved in DNA double-strand break repair. J Biol Chem 2003; 278: 19286–19291.
Chen J, Gomes AR, Monteiro LJ, Wong SY, Wu LH, Ng TT et al. Constitutively nuclear FOXO3a localization predicts poor survival and promotes Akt phosphorylation in breast cancer. PLoS One 2010; 5: e12293.
Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res Treat 2010; 123: 725–731.
Delacote F, Han M, Stamato TD, Jasin M, Lopez BS . An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res 2002; 30: 3454–3463.
Weinstock DM, Brunet E, Jasin M . Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat Cell Biol 2007; 9: 978–981.
Neri B, Pacini P, Bartalucci S, Moroni F, Menchi I, Cappellini M . Epirubicin high-dose therapy in advanced breast cancer: preliminary clinical data. Epirubicin as a single agent in breast cancer. Int J Clin Pharmacol Ther Toxicol 1989; 27: 388–391.
Stiff T, Reis C, Alderton GK, Woodbine L, O'Driscoll M, Jeggo PA . Nbs1 is required for ATR-dependent phosphorylation events. EMBO J 2005; 24: 199–208.
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009; 11: 973–979.
Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7: 126–136.
Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P et al. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J 2006; 25: 5775–5782.
Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M . Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol 2006; 409: 524–540.
Millour J, Constantinidou D, Stavropoulou AV, Wilson MS, Myatt SS, Kwok JM et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010; 29: 2983–2995.
Kang J, Ferguson D, Song H, Bassing C, Eckersdorff M, Alt FW et al. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol 2005; 25: 661–670.
McGovern UB, Francis RE, Peck B, Guest SK, Wang J, Myatt SS et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther 2009; 8: 582–591.
Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW . Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther 2008; 7: 2022–2032.
Nowak DE, Tian B, Brasier AR . Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 2005; 39: 715–725.
Acknowledgements
EW-F Lam, AR Gomes, RC Coombes were supported by grants from Cancer Research UK, EW-F Lam and S Zona by grants from Breast Cancer Campaign, LJ Monteiro grants from Fundação para a Ciência e a Tecnologia, and P Khongkow, M Khongkow and M Kongsema from the Royal Thai Government Scholarships. We also thank Julie Millour for initiating the study and help with this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Khongkow, P., Karunarathna, U., Khongkow, M. et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 33, 4144–4155 (2014). https://doi.org/10.1038/onc.2013.457
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2013.457
Keywords
This article is cited by
-
The forkhead box M1 (FOXM1) expression and antitumor effect of FOXM1 inhibition in malignant rhabdoid tumor
Journal of Cancer Research and Clinical Oncology (2021)
-
MicroRNA-320d regulates tumor growth and invasion by promoting FoxM1 and predicts poor outcome in gastric cardiac adenocarcinoma
Cell & Bioscience (2020)
-
Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing
Nature Communications (2020)
-
FZD5 contributes to TNBC proliferation, DNA damage repair and stemness
Cell Death & Disease (2020)
-
Long noncoding RNA TRPM2-AS acts as a microRNA sponge of miR-612 to promote gastric cancer progression and radioresistance
Oncogenesis (2020)
