
Abstract
High-risk human papillomaviruses are causative agents of cervical cancer. Viral protein E7 is required to establish and maintain the pro-oncogenic phenotype in infected cells, but the molecular mechanisms by which E7 promotes carcinogenesis are only partially understood. Our transcriptome analyses in primary human fibroblasts transduced with the viral protein revealed that E7 activates a group of mitotic genes via the activator B-Myb-MuvB complex. We show that E7 interacts with the B-Myb, FoxM1 and LIN9 components of this activator complex, leading to cooperative transcriptional activation of mitotic genes in primary cells and E7 recruitment to the corresponding promoters. E7 interaction with LIN9 and FoxM1 depended on the LXCXE motif, which is also required for pocket protein interaction and degradation. Using E7 mutants for the degradation of pocket proteins but intact for the LXCXE motif, we demonstrate that E7 functional interaction with the B-Myb-MuvB complex and pocket protein degradation are two discrete functions of the viral protein that cooperate to promote acute transcriptional activation of mitotic genes. Transcriptional level of E7 in patient’s cervical lesions at different stages of progression was shown to correlate with those of B-Myb and FoxM1 as well as other mitotic gene transcripts, thereby linking E7 with cellular proliferation and progression in cervical cancer in vivo. E7 thus can directly activate the transcriptional levels of cell cycle genes independently of pocket protein stability.
Similar content being viewed by others


Introduction
The high-risk human papillomavirus (HPV) E7 protein is multi-functional and influences cellular fitness, metabolic processes, cytokine signalling and mitotic activity in infected cells.1 Efficient modulation of the cell cycle by HPV E7 is likely to be a critical pro-oncogenic event in virus-infected cells. Expression of E6 and E7 in cervical carcinoma cells is both necessary and sufficient to maintain the transformed phenotype, which is subsequently lost upon suppression of E6/E7 transcription.2, 3, 4 E7 directly interacts with and promotes the degradation of pocket proteins (negative regulators of the cell cycle) to allow E2F activators to stimulate S-phase gene transcription and drive host cell mitotic entry.1 Accordingly, suppression of E6 and E7 transcription in cervical carcinoma cells leads to a corresponding transcriptional suppression of S-phase and mitotic genes.4, 5, 6, 7
Two prominent transcriptional activators of mitotic genes, B-Myb and FoxM1, are E2F target genes that are substantially modulated by suppression of E6/E7 transcription in HeLa cells.7 The HPV16 E7 protein had previously been shown to specifically activate B-Myb transcription8 and to interact with and activate FoxM1.9 Mammalian B-Myb is a product of the MYBL2 gene that is expressed in all dividing cells, such that loss of function mutations in this gene causes early embryonic lethality.10 Orthologous MYBL2 in both drosophila11, 12, 13 and zebra fish14 is similarly essential for the regulation of mitotic progression. The mammalian MuvB core acts in conjunction with B-Myb to activate a range of mitotic genes15, 16, 17, 18, 19 and B-Myb has been shown to be activated in cancers, reviewed in.20 The MuvB core itself contains five distinct subunits, among which LIN9 is known to be essential for embryonic development. In contrast, FoxM1 has long been implicated in the execution of the mitotic program in mammalian cells,21, 22 but the interplay between this transcription factor and B-Myb-MuvB complex was only very recently identified.23, 24 FoxM1 influences a plethora of different cellular activities, including regulation of oxidative stress, repair of DNA damage and even tumour metastasis.25, 26 Accordingly, overactive FOXM1 has a major role in many cancers, including breast, lung, colorectal and cervical carcinomas.27, 28, 29, 30, 31 There are two distinct forms of DREAM complexes reported to date in the literature, the B-Myb-MuvB activator of mitotic genes as described above and a repressor complex that will be called DREAM/p130 as it is associated with the p130 pocket protein. The DREAM/p130 complex is involved in repression of S-phase genes in the G0 phase of the cell cycle via interaction with the pocket proteins p130 and p107 and the E2F5 or E2F4 repressors.16, 32 It has recently been shown that E7, by degrading p130, induces activation of S-phase genes through inactivation of the DREAM/p130 repressor complex.33 In contrast to the DREAM/p130 repressor, the B-Myb-MuvB activator does not interact with pocket proteins but rather with transcriptional activators B-Myb and FoxM1. In the present report, we hypothesized that HPV protein E7 can activate mitotic genes in host cells by directly influencing the activity of this B-Myb-MuvB activator complex, independent of pocket proteins. We report that E7 interacts with the main component of the MuvB core, LIN9, and with transcription factors B-Myb and FoxM1 to promote mitosis in infected cells. E7 was recruited to promoters of mitotic genes in vivo in a B-Myb dependent manner. Interaction with LIN9 and FoxM1 depended on the LXCXE motif of E7, whereas E7 interaction with B-Myb did not. However, this novel trans-activating property of E7 was not sufficient to achieve full mitotic activation in primary human cells, unless coupled with depletion of pocket proteins. Our data reveal a novel role for E7 in promoting mitotic genes activation both in vitro and in patient’s clinical samples, thereby functionally complementing the degradation of pocket proteins to enhance host cell proliferation in cervical cancer.
Results
Acute HPV16 E7 expression activates mitotic gene transcript levels in primary human fibroblasts (PHFs)
We sought to compare modulation of cell cycle genes occurring in Caski cells, a cervical carcinoma cell line associated with HPV16, with gene modulation occurring in HPV16 E7 expressing cells. A total of 360 genes were suppressed in Caski cells expressing the transcriptional repressor E2C, which represses endogenous transcription of the HPV16 E6 and E7 oncogenes as previously shown in the HPV18-associated HeLa cells,2, 3, 4 including 134 mitotic genes (Supplementary Table S1). We used the Illumina platform to conduct transcriptome analyses of PHFs after 72 h of viral transduction with HPV16 E7 (in condition of acute E7 expression in most cells). A total of 213 modulated genes were shared between E2C-expressing Caski cells (repressed genes) and E7-expressing fibroblasts (activated genes), including 106 mitotic genes, thus further indicating that E7 is a potent pro-mitotic factor in cervical carcinoma cells and when overexpressed in primary cells (Figure 1a and Supplementary Table S2).

HPV16 E7 mutants that are unable to degrade pocket proteins failed to activate cell cycle genes transcript levels. (a) Microarray analyses were performed to measure gene expression after transcriptional repression of HPV16 E6 and E7 (16E6 and 16E7) by the E2C repressor in Caski cells. A total of 360 genes were down-modulated (>2-fold) by 16E6/E7 repression compared with control cells, see Supplementary Table S1 for the complete list of genes suppressed in E2C-infected Caski cells. Microarray analyses of PHFs at 72 h post transduction with 16E7 were compared with control cells and showed that a total of 975 genes were activated >2-fold by E7 on four replica at P<0.01. Venn diagram of genome-wide expression profiles of 16E7-depleted Caski cells and 16E7-transduced PHFs showing an overlap of 213 genes, among which 126 are cell cycle genes and 106 are mitotic genes analysed by Ingenuity Pathway studio. See also Supplementary Table S2 for list of common S-phase and mitotic genes. (b) Wild-type HPV16 E7 and mutant D6−10 interact with endogenous pRB, but not mutant D21−24. 293 cells were transfected with expression vectors of Flag-tagged wt 16E7, mutant D6−10 or mutant D21−24. CoIP was performed using an anti-Flag antibody, although the western blots were revealed with an anti-pRB antibody. Flag-RFP was used as a negative control. (c) The CT values of HPV16 E7 in Caski cells or in PHFs expressing empty vector (control), wt 16E7, mutant D6−10 or mutant D21−24 were assessed by qPCR. (d) Cells were collected for real-time analyses of a panel of mitotic genes (upper panel) and S-phase genes (lower panel) selected from the microarrays, after 72 h transduction with empty vector, wt16E7 or mutant D6−10 or D21−24. Data shown are derived from three independent experiments and are presented as the mean values of relative mRNA expression±s.e.m. (e) Western blot measurement of protein levels of wild type HPV16 E7 (16E7) and E7 mutants D6−10 and D21−24 in transduced fibroblasts alongside protein levels of the corresponding endogenous mitotic gene CDC25C and S-phase genes FOXM1, MYBL2, LIN9.
We then studied transcription of a subset of these genes found modulated in microarrays by inducing the expression of HPV16 E7 wild type and E7 variants in primary human skin fibroblasts. The E7 variants were mutant E7 D21−24 that lacks the LXCXE motif and thus cannot interact and degrade pocket proteins, or mutant E7 D6−10 that can interact with pocket proteins but cannot degrade them as previously shown34 and in Figures 1b and e. Efficient E7 expression was verified by RT-PCR analyses of transcript levels in infected cells, and the expression conditions were optimized to mimic endogenous levels of expression observed in HPV16-associated cervical carcinoma cell lines Caski and SiHa cells (where E7 transcripts are expressed at ∼14–16 cycles thresholds, CT) (Figure 1c). Levels of the E7 proteins were more difficult to compare due to deficient epitope recognition of the deleted proteins by the antibodies as noted earlier35 (Figure 1e). We were, however, confident that the mutants and wild type E7 were similarly expressed as their flagged counterparts appeared expressed at comparable levels (Figure 1b). Overexpression of wild type E7 induced 8−20-fold upregulation of the transcript levels of six endogenous mitotic genes, whereas neither E7 mutant was able to elicit detectable gene activation (Figure 1d, upper panel). These findings were confirmed by western blots analyses demonstrating that only wild type E7 was capable of increasing the expression of the mitotic protein cdc25C (Figure 1e). When the effect of E7 expression on endogenous proteins in PHFs was examined by western blot, clear increase of B-Myb, FoxM1 and LIN9 endogenous expression could be seen, as well as degradation of pRB in the cells expressing wild-type E7 only (Figure 1e). Wild type E7 also upregulated the transcript levels of three of four representative S-phase genes by ∼5−10-fold, and CCNE2 gene activity was increased ∼70-fold (Figure 1d lower panel), whereas neither E7 mutant was able to activate S-phase genes efficiently. We, therefore, concluded that E7 can comparably stimulate M- and S-phase gene transcript levels in PHFs, whereas the E7 mutants cannot, supporting previously published data on the functional role of degradation of pocket proteins.4
HPV16 E7 cooperates with B-Myb or FoxM1 to increase mitotic gene transcript levels in PHFs
We next investigated whether E7 was capable of activating M-phase genes via effects on the B-Myb-MuvB activating complex. Wild type E7 or mutant E7 proteins were coexpressed with the B-Myb and FoxM1 transcriptional activators of the B-Myb-MuvB complex and the protein levels were assessed by western blot in transduced fibroblasts (Figure 2a). As noted earlier, their function can only be studied using overexpression as primary fibroblasts express very low endogenous levels of both B-Myb and FoxM1 (Figures 1e and 2a). We noted that overexpression of B-Myb decreased pocket protein p130 but we have no explanation for this phenomenon at the moment. Transduction of MYBL2 or E7 alone into PHFs increased transcription of a representative panel of three endogenous mitotic genes that were chosen from the microarrays data (Supplementary Data Table S1) (CDC25C, PLK4 and SPC25) and coexpression of both E7 and B-Myb induced further increase (Figure 2b) that was less obvious for the S-phase genes (CCN2, POLA2 and TK1) (Figure 2d). We next examined the effect of B-Myb coexpression with mutant E7 proteins. Although E7 D6−10 did not activate transcription of the mitotic genes as shown previously (Figure 1d), coexpression with B-Myb induced a strong cooperative increase in transcript levels (3−5-fold compared with B-Myb alone) of the three mitotic genes (Figure 2c) as well as of three representative S-phase genes (Figure 2e). Increase in the transcript levels of the selected mitotic and S-phase genes was also observed in cells that expressed FoxM1, and coexpression of both FoxM1 and E7 D6−10 induced a cooperative increase of M-phase gene transcript levels (Supplementary Figure S1A). Functional cooperation between B-Myb or FoxM1 and E7 D6−10, leads to levels of mitotic genes transcription that reach the wild type E7 cooperative levels, thus strongly suggesting that this functional cooperation is indeed independent of degradation of pocket proteins. Interestingly, when the same experiments were done with the E7 LXCXE mutant, less cooperativity was seen indicating that the LXCXE motif may have a role in E7 cooperation with B-Myb and FoxM1 and the B-Myb-MuvB complex (Supplementary Figure S1A and S1B).

Coexpression of HPV16E7 with B-Myb induces cooperative activation of mitotic genes transcript levels in PHFs. (a) Western blot analyses showing expression of 16E7, B-Myb and FoxM1 and concomitant degradation of endogenous p130 and pRB in wild type E7 transduced skin PHFs. (b) PHFs were transduced with B-Myb and wild type 16E7 (c) same as (b) but with E7 mutant D6−10. After 72 h, fibroblasts were collected and subjected to qPCR analyses using specific primers for mitotic genes: CDC25C, PLK4 and SPC25. (d) Same as (b) but the qPCR analyses were done with primers for the S-phase genes: CCNE2, POLA2 and TK1 as indicated. (e) Same as (c) for the S-phase genes. Results shown are representative of three or more independent experiments.
HPV16 E7 interacts with B-MyB and FoxM1 and is recruited to mitotic promoters
The synergistic increase of mitotic genes transcript levels by HPV16 E7 and B-Myb or FoxM1 strongly suggested that E7 directly interacts with B-Myb or FoxM1, as demonstrated previously for FoxM1 only,9 and could perhaps form transcriptional complexes with these proteins. We, therefore, looked for E7:B-Myb interaction by performing immunoprecipitation in extracts of 293T cells that overexpressed E7 and B-Myb and were indeed able to detect an interaction between these two proteins (Figure 3a). The E7:B-Myb interaction was largely preserved when these assays were repeated using the E7 LXCXE mutant (D21−24), and were completely preserved in analyses of the D6−10 mutant, indicating that binding of B-Myb to E7 is independent of the LXCXE motif (Figure 3a). Wild type E7 and the D6−10 mutant both interacted strongly with FoxM1, but unlike E7: B-Myb interaction, deletion of the LXCXE motif substantially reduced E7 interaction with FoxM1 (Figure 3b). As p130, has been shown to interact with both the DREAM/p130 repressor complex and E7, we wanted to make certain that interaction of E7 with B-Myb-MuvB did not occur via pocket proteins. The same coimmunoprecipitation experiments, performed in cells where pocket proteins were silenced by use of small hairpin (sh)RNAs as described,36 gave rise to levels of interaction comparable as in untreated cells (Supplementary Figure S2). This experiment thus firmly establishes that E7 interacts with the B-Myb and FoxM1 independently of the pocket proteins interaction and/or degradation despite the partial requirement for the LXCXE motif. To further clarify the mechanism by which E7 regulates mitotic genes, we performed chromatin immunoprecipitation experiments that focused on the regulatory regions of mitotic genes (AURKB, CDC25B, ESPL1 and SPC25) in cells that overexpressed a tagged HPV16 E7 protein. These assays confirmed that E7 is actively recruited to the promoters of mitotic genes in HeLa cells, although the level of recruitment was weak (Figure 3c). As transcription factors, B-Myb and FoxM1, were capable of binding to specific sequences in mitotic gene promoters and could thus potentially mediate E7 interactions with chromatin, we performed sequential chromatin IP in PHFs expressing E7 alone or E7 coexpressed with B-Myb. Recruitment of E7 to mitotic promoters was detected only in the presence of coexpressed B-Myb in a complex containing LIN54, one of the element of MuvB core, therefore unambiguously demonstrating that E7 is actively recruited to mitotic genes promoters through the B-Myb-MuvB complex (Figure 3d). In addition, these experiments show that an elevated B-Myb expression is required for E7 to be recruited to promoters as recruitment of E7 alone could only be seen in HeLa cells and not in fibroblasts where endogenous B-Myb is very low.

HPV16 E7 interacts with B-Myb and FoxM1 and binds to mitotic gene promoters. (a) Protein extracts from 293T cells expressing Flag-tagged 16E7, D21−24 or D6−10 and B-Myb proteins were immunoprecipitated with Flag antibodies and then analysed by western blot using the B-Myb antibody. Input controls comprised 2% of the lysates used for immunoprecipitation. B-Myb can associate with wild-type 16E7 as well as with mutants D21−24 and D6−10. (b) Same experiment as in (a) but with coexpression of 16E7 and FoxM1, which interacts with wt 16E7 and D6−10 but less markedly with D21−24. Flag-tagged RFP was used as a negative control. (c) ChIP assays were performed using anti-Flag antibody with extracts from HeLa cells ectopically expressing Flag-tagged-16E7. 16E7 was found to occupy the promoters of several mitotic genes: AURKB, CDC25B, ESPL1 and SPC25. Relative occupancy values were calculated by determining the levels of immunoprecipitated DNA compared with that of the input samples and were normalized to that of GAPDH. Histogram depicts fold enrichment of promoter sequences in immunoprecipitates with anti-flag antibodies compared with IgG pull-down samples. Data depicted represent the mean enrichment values±s.d. P-values were calculated using Student’s paired t-test: *P⩽0.05. (d) E7-LIN54 sequential ChIP assays were performed on human fibroblasts cells transduced with Flag-tagged 16E7 alone or together with B-Myb. Complexes eluted from primary ChIP using anti-Flag antibodies were incubated overnight with anti-LIN54 antibodies. Values reported in the graph are fold enrichment of promoter sequences after normalization with samples pull-down with IgG followed by anti-LIN54 antibody.
HPV16 E7, B-Myb and FoxM1 interact with the LIN9 component of the MuvB core
B-Myb has previously been shown to activate the transcription of mitotic genes by interacting with the LIN9 member of the mammalian MuvB core.17, 18, 19 Accordingly, we were able to confirm that B-Myb interacts with LIN9 when coexpressed in human fibroblasts as well as an interaction of LIN9 with FoxM1 (Figure 4a). In immunoprecipitation experiments, we could also detect an interaction between E7 and LIN9, although this interaction was markedly reduced if E7 lacked the LXCXE motif (Figure 4b). In contrast, the D6−10 region of E7 was dispensable for interaction with LIN9. Further analyses also revealed a lower−strength interaction between FoxM1 and B-Myb (Figure 4c). These data and the above ChIP experiments strongly suggested that B-Myb, FoxM1 and HPV16 E7 are part of the B-Myb-MuvB transcriptional activator complex that drives mitotic gene transcription. At that point we wanted to clarify whether E7 activation of cell cycle genes observed in fibroblasts was indeed dependent on B-Myb-MuvB and could demonstrate that B-Myb knockdown decreased E7-dependent activation of cell cycle genes transcript levels (Figure 4d).

HPV16 E7, B-Myb and FoxM1 interact with the LIN9 component of the MuvB core and E7-dependent increase of cell cycle gene transcripts is dependent on B-Myb. (a) Coimmunoprecipitation assays were conducted in 293T cells co-transfected with GFP-LIN9 and Flag-tagged FoxM1 or Flag-tagged B-Myb (positive control) or Flag-tagged RFP (negative control). (b) Same assays as in (a) but conducted in cells co-transfected with 16E7, D21−24 or D6−10, showing that both E7 wild type and mutant D6−10 interact with LIN9, whereas this interaction is reduced for the mutant D21−24. (c) Proteins were extracted from 293T cells co-transfected with Flag-tagged B-Myb and FoxM1 before being subjected to immunoprecipitation using anti-Flag antibodies. (d) Efficiency of the B-Myb knock down by siRNA transduction in PHFs (left graph) and cell cycle gene transcript levels in control and E7 transduced fibroblasts in the presence of the B-Myb shRNA (right graphs). PHFs were simultaneously transduced by viruses carrying 16E7 or empty vector and siRNA against B-Myb for 3 days before collecting for qPCR analysis.
E7 interaction with the B-Myb-MuvB complex is complemented by pocket protein degradation to fully enhance cell cycle genes transcription
We transduced shRNAs targeting pocket proteins pRb, p130 or p107 into primary human skin fibroblasts and evaluated the depletion of the corresponding proteins (Figure 5a).36, 37 If the pocket proteins were silenced separately, none of these depletion alone could induce detectable increase in cell cycle genes expression (Supplementary Figure S3A (−) lanes). When coexpressed with wt E7, depletion of the pocket proteins had only very little effect on the wt E7 gene activation. In contrast, pRB depletion gave rise to a detectable increase in cell cycle genes transcript levels when coexpressed with E7 D6−10 and at a much lower levels than wt E7 for the mitotic genes (Supplementary Figure S3A), although it was more sustained for the S-phase genes (Supplementary Figure S3B). We thus decided to deplete the three pocket proteins together and cotransduced the three shRNA to achieve ∼80% silencing of pRb and p130 alongside the previously noted lower efficiency silencing of p107 in cosilencing experiments probably owing, at least in part, to p107 being activated to compensate for loss of pRB37 (Figure 5b). Further increase in transcript levels of cell cycle genes was observed after silencing of the two pocket proteins p130 and pRB; mitotic genes were upregulated 1−2-fold, whereas S-phase genes were upregulated ∼5−20-fold (control columns (−) in Figures 5c and d). Wild type or mutant E7 proteins expression alongside silencing of p130 and pRB led to a slight increase in mitotic genes transcription by wild type E7, although it remarkably restored the lack of activity of the D6−10 mutant to levels 50 to 80% of wild type E7 (Figure 5c). In stark contrast, the pro-mitotic activity of the D21−24 mutant was not rescued by partial depletion of pocket proteins (Figure 5c), thus confirming that another function of E7 LXCXE motif is involved here such as interaction with LIN9 or FoxM1 of the B-Myb-MuvB activator complex, as demonstrated earlier (Figure 3b and 4b). A similar cooperative activation between E7 D6−10 and silencing of pocket proteins was seen for S-phase genes activation with almost full recovery of the wild type E7 activity (Figure 5d).

Pocket protein degradation complements E7:B-Myb-MuvB interaction in E7-induced increase of cell cycle genes transcription. (a) Western blots showing knock down efficiency of individual pocket protein silencing in fibroblasts transduced with the corresponding shRNAs against pRB (shpRB), p107 (shp107) and p130 (shp130) (b) Knockdown efficiency of pocket protein transcription in PHFs transduced with the three pocket proteins shRNAs together were determined by qPCR. (c) PHFs were transduced with either E7 wild type or mutants D6−10 or D21−24 as indicated at the bottom of the graphs, (−) indicating cells transduced with the empty pCDH vector, together with gene silencing of pocket proteins by lentiviral delivery of shRNAs. These cells were assayed for expression of three M-phase genes using qPCR after 72 h coinfection. (d) Same experiments as in (c) but assayed with three S-phase genes. Data shown are derived from three independent experiments and are presented as the mean mRNA values±s.e.m.
The complementary functions of E7 in primary human keratinocytes
As the natural host for HPV infection and E7 cellular transformation is the human keratinocyte, we validated our fibroblast data by conducting an additional series of experiments in primary human keratinocytes. Transduction of shRNAs against pocket proteins into human keratinocytes efficiently suppressed the transcription of pRb and p130, and weakly suppressed p107 (Figure 6a), but activation of endogenous M-phase genes was not detected in this condition (Figures 6b and c). In contrast, transduction of wild type E7 into primary human keratinocytes induced substantial activation of mitotic genes (Figure 6b) and S-phase genes (Figure 6c). Western blot analyses confirmed that expression of wild type E7 increased the protein expression of cell cycle genes CDC25C, MAD2 and CCNE1, although partial silencing of pocket proteins was unable to activate them (Figure 6d). A model of enhanced activation of mitotic genes by E7 is shown, where E7 can activate the cell cycle genes by two cooperating pathways and a strong feed forward loop (Figure 6e).

Acute expression of HPV16 E7 activates cell cycle genes transcriptional levels in primary human keratinocytes (PHKs) more efficiently than pocket proteins silencing. (a) Real-time PCR analyses were performed to verify gene silencing efficiency of lentiviral shRNA targeting of pRB, p107 and p130 together in PHKs. (b) PHKs transduced with shRNAs against pocket proteins (pRB, p107 and p130) and with HPV16E7 were collected for real-time PCR analyses of six M-phase genes. (c) Same experiment as in (b) but analysing transcription levels of five S-phase genes. Results shown are representative of two independent experiments conducted on two different batches of PHKs. (d) Western blot analyses of 16E7, CDC25C, MAD2 and CCNE1 expression in PHKs after gene silencing of pocket proteins for comparison with over-expression of 16E7. *indicates a non-specific band. (e) Model of mitotic genes activation in the presence or absence of E7.
Correlation between E7 and mitotic genes transcriptional modulation in HPV16-associated patients cervical dysplasia
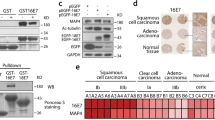
We next investigated transcript levels of E7 and cell cycle genes in patient clinical samples, by using NanoString technology.38, 39 Transcript levels of six M- and two S-phase genes were analysed in cervical biopsy extracts from three healthy controls, eight patients with grade 2−3 cervical intraepithelial neoplasia and four patients with squamous cell carcinoma. For comparison, we performed parallel analyses of three cervical carcinoma cell lines previously shown to be associated with HPV16: Caski, SiHa and IC3, this last cell line being described in.40 E7 transcript levels were compared with the transcript levels of a series of mitotic genes identified by the microarrays described above (Supplementary Table S1). HPV16 E7 expression levels in biopsy extracts were correlated with MYBL2 and FOXM1 genes and with mitotic genes CCNB1, AURKB, PLK1, CDC20, CDC25C and CENPA (r>0.84; Figure 7), thus confirming a relationship between E7 expression and mitotic gene activation in human lesions. In validation of our in vitro systems, E7 expression was also shown to correlate with cellular mitotic genes in the cervical carcinoma cell lines Caski, SiHa and IC3 and E7 transcription levels were comparably decreased in Caski cells expressing the HPV18 E2C repressor (Figure 7). We concluded, therefore, that E7 transcript levels in cervical biopsy samples correlate with transcript levels of mitotic genes, which supports a direct role for HPV E7 in modulating these genes in HPV-infected cells in vivo. Activation of mitotic genes was detected from low-grade cervical intraepithelial neoplasia 2 lesion indicating that E7 is able to activate mitotic genes at the early stages of transformation, but that transcription of the viral gene increases correlatively with activation of the host mitotic genes and grade of the clinical lesions.

Transcriptional activation of MYBL2, FOXM1 and mitotic genes correlates with increase of HPV16 E7 transcripts in human biopsy samples and cervical carcinoma cells. Human biopsy RNA samples including three normal cervix (black), eight cervical intraepithelial neoplasia (Blue) and four squamous cell carcinoma (red) associated with HPV16 were subjected to transcriptome analysis using the NanoString technology. In parallel, cervical carcinoma cell lines SiHa, Caski and IC3 (brown) and Caski cells expressing exogenous E2 (yellow) were analysed using the same approach. Levels of HPV16 E7 expression were compared with levels of mitotic gene transcripts, as indicated on the panels of the Figure. Expression of each of the mitotic genes analysed was significantly correlated with E7 expression. Transcription levels of both E7 and mitotic genes increased substantially with increasing lesion severity.
Discussion
We demonstrate here that incorporation of E7 into the activating B-Myb-MuvB complex promotes increase in mitotic gene transcript levels, but is not sufficient in primary cells as this process must be complemented by silencing of pocket proteins to achieve full effect. We also demonstrate that pocket proteins depletion does not recapitulate E7 expression to activate cell cycle genes in primary cells. Importantly, our experiments were done in acute condition where E7 is expressed transiently in ∼100% transduced cells, and genes were analysed in a short time frame of 2−3 days. In similar transduction experiments, pocket proteins shRNAs were also delivered to ∼100% of the cells and the reduction in pRb and p130 expression was ∼80%. The lack of strong cell cycle genes activation through pocket protein silencing in our system, both in fibroblasts and keratinocytes in monolayers, indicates that more time is needed for the genes to be activated because that activation occurs after a cascade of events. RB depletion is comparable with E7 expression in inducing cell proliferation only in long term experiments involving either raft cultures or mouse models.41, 42, 43 In contrast, E7 activation of the genes is rapid due to direct interaction with the transcriptional activator B-Myb-MuvB and immediate increase in its function. This is amplified by a feed forward loop where E7 activates the B-Myb and FoxM1 transcription factors as well as the MuvB core constitutive element LIN9, as depicted in the model in Figure 6e. There is, therefore, a difference in phenotypes of long term and short term Rb depletion, which has already been noted by others.42 In any case the same authors noted that even though Rb depletion could recapitulate E7 expression in inducing cell proliferation in mice skin, E7 effect is more rapid than Rb depletion. In addition, they reported that expression of E7 together with Rb depletion gave a more pronounced phenotype indicating that E7 exhibits additional functions to Rb degradation. Taken together, all these data led us to the hypothesis that E7 activation of B-Myb-MuvB is used to accelerate the cellular proliferation and to initiate and stimulate a powerful feed forward loop of cell cycle gene activation including S-phase genes, through a transcriptional cooperation between B-Myb-MuvB and E2F. Interestingly, we found that the interaction with the B-Myb-MuvB complex is partially conserved with the low risk HPV6 and 11 E7 proteins, thus pointing to common pro-proliferative mechanisms in the high and low risk models (Toh et al. manuscript in preparation).
The B-Myb-MuvB complex is shown here to be an important new target of oncogenic transformation in human cells, as is also indicated by the overexpression of B-Myb and FoxM1 observed in many cancers. Our data further indicate that the entire B-Myb-MuvB complex (including LIN9) is activated in HPV16-infected lesions and cervical cancer. Consistent with our findings, a recent report from Lambert laboratory demonstrated that E7-mediated degradation of pocket proteins alone is not sufficient to induce tumorigenesis in a mouse model of cervical carcinoma.37 It was also shown that E7 proteins from various HPV types stimulate proliferation independently of their association to pRB.44 We, therefore, propose that B-Myb-MuvB activation is also required for carcinogenic progression. In addition to confirming E7 as a major oncogene in a HPV model, the current study identifies a novel role for E7 in inducing the transcriptional activation of mitotic genes and in accelerating the cell cycle in HPV-infected cells by direct protein−protein interaction with a transcriptional activator complex. As this interaction is probably required for carcinogenic progression in human patients, then therapeutic blockade of this activity could provide new treatment strategies in cervical carcinoma.
Materials and methods
Microarray hybridization and analysis
The Human Gene 1.0 ST array from Affymetrix (Santa Clara, CA, USA), (including 28 869 well-annotated genes with 764 885 distinct probes) was used to analyse E2C-infected Caski cells (Figure 1a). In each experiment, a total of 200 ng RNA was amplified, terminally labelled and used for hybridization according to the manufacturer’s instructions. Data were analysed and tested for statistical significance using Partek Genomics Suite software (St Louis, MO, USA). Illumina Human HT-12 V4 expression BeadChip (Singapore) was employed for genome-wide expression profiling of E7-transduced fibroblasts. Microarrays were performed by the microarray facility at Biopolis Shared Facilities (A-star, Singapore). Genes were analysed by the Ingenuity Pathway Studio (Redwood city, CA, USA).
Cell culture
HeLa, Caski, 293T cells and primary human skin fibroblasts were grown in Dulbecco’s Modified Eagle Medium (Invitrogen, Singapore) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Primary human neonatal foreskin keratinocytes were purchased from CELLnTEC (Scarborough, ON, Canada) and cultured in PCT epidermal keratinocyte medium (CnT-57, CELLnTEC) at 37 °C, in a humidified 5% CO2 incubator.
Expression vectors
Vectors expressing B-Myb or FoxM1B were constructed by amplifying the corresponding DNA fragments from pBABE-B-Myb and pSIN-CMV-FoxM1B, respectively.29 The full-length, wild-type HPV16 E7 and HPV16 E7 mutants D6−10 and D21−24 were subcloned from pLXSN-16E7, pLXSN-16E7D6−10 and pLXSN-16E7D21−2445 into mammalian expression vector pXJ-Flag, whereas the GFP-tagged human LIN9 used in the coimmunoprecipitation experiments was described in.16
Transfection and viral transduction
Microarrays in Caski cells were performed after 48 h of infection with recombinant adenoviruses expressing either GFP-HPV18 E2C or GFP in conditions described previously.6 For transient gene expression or gene knockdown experiments, 293T cells were transfected with Fugene HD (Roche, Singapore, 168730) using the respective plasmid constructs according to the manufacturer’s recommendations. PHFs and keratinocytes were transduced with viral vector carrying B-Myb or FoxM1B and with wt, D6−10 or D21−24 mutants of HPV16 E7 (or empty vector), and were collected 72 h post-infection for real-time PCR to confirm gene expression. For silencing of pocket proteins, infections were done in PHFs and keratinocytes using viruses expressing shRNA against pRB, p107 and p130.36 Small-interfering-RNA duplex was designed for B-Myb: forward 5′-GAUCUGGAUGAGCUGCACUTT-3′ reverse 5′-AGUGCAGCUCAUCCAGAUCTT-3′.
Real-time PCR
RNA was extracted using RNeasy Mini Kit (Qiagen, Singapore) according to the manufacturer's instructions. Reverse transcription was performed as previously described.6 Each quantitative PCR was performed in duplicate for each primer set. Relative transcript amounts were calculated by the ΔCT method using GAPDH as a reference gene and sequences of the primers are given in Supplementary Data Table S3.
Immunoprecipitation, immunoblotting and chromatin immunoprecipitation
293T cells were lysed using ML buffer (300 mM NaCl, 0.5% NP-40, 20 mM Tris-HCl (pH 8.0), 1 mM EDTA) supplemented with one tablet of complete EDTA-free protease inhibitor cocktail (Roche) per 50 ml lysis buffer. Supernatants were collected after centrifugation at 13 000 r.p.m for 20 min. Protein concentrations were determined by the BCA method (Pierce, Thermo Scientific, Tech Park, Singapore). Samples containing 500 μg protein were mixed with 30 μl Protein A/G Agarose beads (Santa Cruz Biotechnology. Inc., Santa Cruz, CA, USA) and 2.5 μg anti-Flag M2 antibody (Sigma-Aldrich, St Louis, MO, USA) and incubated for 5 h at 4 °C under constant rotation. Immune complexes were washed three times in ML buffer and subjected to SDS−PAGE and immunoblot analysis. Blots were probed sequentially with primary and secondary antibodies at the following dilutions: anti-HPV16 E7 at 1:200 (8C9, Zymed, Invitrogen, Singapore) and sc-6981, (Santa Cruz), anti-Flag at 1:2000 (F7425, Sigma-Aldrich), anti-B-Myb at 1:500 (sc-724, Santa Cruz), anti-FoxM1 at 1:1000 (sc-502, Santa Cruz), anti-pRB at 1:1000 (#9309, Cell Signaling Technology. Inc., Danvers, MA, USA), anti-p130 at 1:500 (sc-317, Santa Cruz), anti-Cyclin E1 at 1:500 (NCC-CyclinE, Leica Biosystems, Singapore), anti-cdc25C at 1:1000 (5H9, Cell Signaling), anti-Mad2 at 1:1000 (sc-47747, Santa Cruz) anti-GFP at 1:5000 (TP401, Torrey Pines, Biolabs, Secaucus, NJ, USA), HRP-conjugated anti-Myc at 1:500 (sc-40 HRP, Santa Cruz), anti-mouse and anti-rabbit at 1:10 000 (GE Healthcare, Chalfont St Giles, UK). Proteins were detected by incubation with ECL substrate (Amersham Bioscience, Piscataway, NJ, USA) for 5 min and chemiluminescence was visualized by STORM imaging system (Amersham, Pleasanton, CA, USA). Chromatin immunoprecipitation were performed as previously described.46 For sequential ChIP, complexes from primary ChIP using anti-Flag antibodies (to bind Flagged-16E7) were eluted twice using 10 mM DTT for 20 min at 37 °C, diluted 10 times with re-ChIP buffer (20 mM Tris-HCl pH 8.1, 0.1% Triton X-100, 2 mM EDTA, 150 mM NaCl) followed by incubation with anti-LIN54 antibodies overnight, and then again subjected to the ChIP procedure. RNA Pol II antibody (39097, Active Motif, Carlsbad, CA, USA) was included as a positive control for efficient transcription of genes in every ChIP assay reported here. Promoter primers: GAPDH F5′-GTCTGCCCTAATTATCAGGTCCAG-3′, R5′-AGGTCACGATGTCCTGCAGC -3′; AURKB F5′-GCAACGAAAGGTCTATTGGTG G-3′, R5′-TCTAACTTCTCTGCCCGATGGAG-3′; CDC25B F5′-AAG AGC CCA TCAGTTCCGCTT G-3′, R5′-CCCATTTTACAGACCTGGACG C-3′; ESPL1 F5′-GACTAAGCGACAGGTAGCTGAGAAA-3′, R 5′-TTGCTGCTCTGCGCGTTA-3′; SPC25 F5′-AAAGAACTACGAATCCCAGAATGC-3′, R 5′-TCTCCTTGACAGGATTGGTTGA-3′; CTGF F5′-CGAGGAATGTCCCTGTTTGTGT, R 5′- GGCCCGAGGCTTTTATACG.
Clinical samples and NanoString nCounter analysis
Fifteen formalin-fixed and paraffin-embedded cervical specimens were selected from the archives of the department of Pathology; National University Hospital affiliated to National University of Singapore. The study was approved by the Institutional Review Board of the National University of Singapore (continuing review of NUS-IRB 09-218).
Total RNA was extracted from the formalin-fixed and paraffin-embedded samples using Qiagen’s RNeasy formalin-fixed and paraffin-embedded kit. Gene expression was profiled by the NanoString nCounter Analysis System, Life sciences (Seattle, WA, USA) according to the manufacturer’s instructions and as described previously.39 The color-coded probe sets were designed and synthesized by NanoString Technologies. Color-coded barcodes on the reporter probes were read and quantified by the nCounter Digital Analyser. Nanostring probes sequences are given in Supplementary Table S4.
Accession numbers
FOXM1: 602341, MYBL2: 601415, LIN9: 609375, MAD2L1: 601467, CDC25C: 157680, PLK4: 605031, SPC25: 609395, AURKB: 604970, ESPL1: 604143, pRB: 614041, p107: 116957, p130: 180203, CCNB1: 123836, CCNE1: 123837, CCNE2: 603775, MCM6: 601806, POLA2: 23649, TK1: 188300, CENPA: 117139, CDC20: 603618, GAPDH: 138400.
References
McLaughlin-Drubin ME, Munger K . The human papillomavirus E7 oncoprotein. Virology 2009; 384: 335–344.
Dowhanick JJ, McBride AA, Howley PM . Suppression of cellular proliferation by the papillomavirus E2 protein. J Virol 1995; 69: 7791–7799.
Desaintes C, Demeret C, Goyat S, Yaniv M, Thierry F . Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J 1997; 16: 504–514.
Johung K, Goodwin EC, Dimaio D . Human papillomavirus e7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J Virol 2007; 81: 2102–2116.
Wells SI, Aronow BJ, Wise TM, Williams SS, Couget JA, Howley PM . Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc Natl Acad Sci 2003; 100: 7093–7098.
Teissier S, Ben Khalifa Y, Mori M, Pautier P, Desaintes C, Thierry F . A new E6/P63 pathway, together with a strong E7/E2F mitotic pathway, modulates the transcriptome in cervical cancer cells. J Virol 2007; 81: 9368–9376.
Thierry F, Benotmane MA, Demeret C, Mori M, Teissier S, Desaintes C . A genomic approach reveals a novel mitotic pathway in papillomavirus carcinogenesis. Cancer Res 2004; 64: 895–903.
Lam EW, Morris JD, Davies R, Crook T, Watson RJ, Vousden KH . HPV16 E7 oncoprotein deregulates B-myb expression: correlation with targeting of p107/E2F complexes. EMBO J 1994; 13: 871–878.
Luscher-Firzlaff JM, Westendorf JM, Zwicker J, Burkhardt H, Henriksson M, Muller R et al. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene 1999; 18: 5620–5630.
Tanaka Y, Patestos NP, Maekawa T, Ishii S . B-myb is required for inner cell mass formation at an early stage of development. J Biol Chem 1999; 274: 28067–28070.
Manak JR, Wen H, Van T, Andrejka L, Lipsick JS . Loss of Drosophila Myb interrupts the progression of chromosome condensation. Nat Cell Biol 2007; 9: 581–587.
Fung SM, Ramsay G, Katzen AL . Mutations in Drosophila myb lead to centrosome amplification and genomic instability. Development 2002; 129: 347–359.
Katzen AL, Jackson J, Harmon BP, Fung SM, Ramsay G, Bishop JM . Drosophila myb is required for the G2/M transition and maintenance of diploidy. Genes Dev 1998; 12: 831–843.
Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J et al. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc Natl Acad Sci USA 2005; 102: 13194–13199.
Georlette D, Ahn S, MacAlpine DM, Cheung E, Lewis PW, Beall EL et al. Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev 2007; 21: 2880–2896.
Pilkinton M, Sandoval R, Colamonici OR . Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene 2007; 26: 7535–7543.
Pilkinton M, Sandoval R, Song J, Ness SA, Colamonici OR . Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J Biol Chem 2007; 282: 168–175.
Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B et al. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J 2007; 26: 144–157.
Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle 2007; 6: 1903–1913.
Martinez I, Dimaio D . B-Myb, cancer, senescence, and microRNAs. Cancer Res 2011; 71: 5370–5373.
Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 2005; 7: 126–136.
Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol 2005; 25: 10875–10894.
Down CF, Millour J, Lam EW, Watson RJ . Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. Biochim Biophys Acta 2012; 1819: 855–862.
Sadasivam S, Duan S, Decaprio JA . The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012; 26: 474–489.
Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD et al. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res 2012; 72: 655–665.
Alvarez-Fernandez M, Halim VA, Krenning L, Aprelia M, Mohammed S, Heck AJ et al. Recovery from a DNA-damage-induced G2 arrest requires Cdk-dependent activation of FoxM1. EMBO Rep 2010; 11: 452–458.
Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D et al. Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res 2009; 69: 3501–3509.
Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res 2006; 66: 2153–2161.
Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, Quinn AG . FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res 2002; 62: 4773–4780.
Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev 2004; 18: 830–850.
Chan DW, Yu SY, Chiu PM, Yao KM, Liu VW, Cheung AN et al. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis. J Pathol 2008; 215: 245–252.
Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007; 26: 539–551.
Nor Rashid N, Yusof R, Watson RJ . Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J Gen Virol 2011; 92: 2620–2627.
Phelps WC, Munger K, Yee CL, Barnes JA, Howley PM . Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J Virol 1992; 66: 2418–2427.
Psyrri A, DeFilippis RA, Edwards AP, Yates KE, Manuelidis L, DiMaio D . Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res 2004; 64: 3079–3086.
Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 2000; 14: 3037–3050.
Shin MK, Sage J, Lambert PF . Inactivating all three rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res 2012; 72: 5418–5427.
Xue Y, Lim D, Zhi L, He P, Abastado JP, Thierry F . Loss of HPV16 E2 protein expression without disruption of the E2 ORF correlates with carcinogenic progression. Open Virol J 2012; 6: 163–172.
Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008; 26: 317–325.
Peter M, Stransky N, Couturier J, Hupe P, Barillot E, de Cremoux P et al. Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol 2010; 221: 320–330.
Ueno T, Sasaki K, Yoshida S, Kajitani N, Satsuka A, Nakamura H et al. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 2006; 25: 4155–4164.
Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF . Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol 2003; 23: 9094–9103.
Buitrago-Perez A, Hachimi M, Duenas M, Lloveras B, Santos A, Holguin A et al. A humanized mouse model of HPV-associated pathology driven by E7 expression. PLoS One 2012; 7: e41743.
Caldeira S, de Villiers EM, Tommasino M . Human papillomavirus E7 proteins stimulate proliferation independently of their ability to associate with retinoblastoma protein. Oncogene 2000; 19: 821–826.
Halbert CL, Demers GW, Galloway DA . The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol 1991; 65: 473–478.
Teissier S, Pang CL, Thierry F . The E2F5 repressor is an activator of E6/E7 transcription and of the S-phase entry in HPV18-associated cells. Oncogene 2010; 29: 5061–5070.
Acknowledgements
We thank Dr DeCaprio for the generous gift of LIN9 and LIN54 antibodies and Birgit lane’s lab for providing the primary human fibroblasts. We also thank Dr Perou for plasmid pBABE-B-Myb; Dr Teh for plasmid pSIN-CMV-FoxM1B; Dr Galloway for plasmids pLXSN-16E7, pLXSN-16E7D6−10 and pLXSN-16E7D21−24; Dr Manser for plasmid pXJ-Flag; Dr Colamonici for plasmid pEGFP-C3-LIN9; Dr Roman for plasmid pHA-p130; Dr Sage for shRNA vectors for pRB, p107 and p130. We would like to express our thanks to Dr Diana Lim from National University Hospital for providing us with the clinical specimens, Drs Zhi Liang and Jean-Pierre Abastado for help in the nanostring experiment design and analyses and John Connolly from SIgN Immunomonitoring Core. We would like to express our thanks to Dr Philipp Kaldis for valuable suggestions. We also greatly appreciate the careful reading of the manuscript by Dr Neil McCarthy of Insight Editing London. This work was supported by Biomedical Sciences Institutes of A-star, Singapore and Chai Ling Pang was a scholar of the National University of Singapore Graduate School for Integrative Science and Engineering.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
Conceived and designed the experiments: CLP, SYT, FT. Performed the experiments: CLP, SYT, PH, YBK, YX. Analysed the data: CLP, SYT, ST, FT. Wrote the paper CLP, FT.
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Pang, C., Toh, S., He, P. et al. A functional interaction of E7 with B-Myb-MuvB complex promotes acute cooperative transcriptional activation of both S- and M-phase genes. (129 c). Oncogene 33, 4039–4049 (2014). https://doi.org/10.1038/onc.2013.426
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2013.426
Keywords
This article is cited by
-
MPS1 is involved in the HPV16-E7-mediated centrosomes amplification
Cell Division (2021)
-
WDHD1 facilitates G1 checkpoint abrogation in HPV E7 expressing cells by modulating GCN5
BMC Cancer (2020)
-
MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis
Cell Death & Disease (2017)
-
Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway
Scientific Reports (2017)
-
Deregulation of the FOXM1 target gene network and its coregulatory partners in oesophageal adenocarcinoma
Molecular Cancer (2015)

{kind=link}
{kind=link}
{kind=link}