
Abstract
Inflammation can act as a crucial mediator of epithelial-to-mesenchymal transition (EMT). In this study, we show that oncostatin M (OSM) is expressed in an autocrine/paracrine fashion in invasive breast carcinoma. OSM stimulation promotes spontaneous lung metastasis of MCF-7 xenografts in nude mice. A conspicuous epigenetic transition was induced by OSM stimulation not only in breast cancer cell lines but also in MCF-7 xenografts in nude mice. The expression of miR-200 and let-7 family members in response to OSM stimulation was downregulated in a signal transducer and activator of transcription factor 3 (Stat3)-dependent manner, resulting in comprehensive alterations of the transcription factors and oncoproteins targeted by these microRNAs. Inhibition of Stat3 activation or the ectopic expression of let-7 and miR-200 effectively reversed the mesenchymal phenotype of breast cancer cells. Stat3 promotes the transcription of Lin-28 by directly binding to the Lin-28 promoter, resulting in the repression of let-7 expression and concomitant upregulation of the let-7 target, high-mobility group A protein 2 (HMGA2). Knock down of HMGA2 significantly impairs OSM-driven EMT. Our data indicate that downregulation of let-7 and miR-200 levels initiates and maintains OSM-induced EMT phenotypes, and HMGA2 acts as a master switch of OSM-induced EMT. These findings highlight the importance of Stat3-coordinated Lin-28B–let-7–HMGA2 and miR-200–ZEB1 circuits in the cytokine-mediated phenotypic reprogramming of breast cancer cells.
Similar content being viewed by others


Introduction
Proinflammatory cytokines have an important role in creating an inflammatory microenvironment, which is characterized as the seventh hallmark of cancer.1 The importance of inflammatory mediators in epithelial-to-mesenchymal transition (EMT), a driver of invasion and metastasis of cancer, is increasingly being recognized.2 Examples of such cytokines in solid tumors include transforming growth factor (TGF)-β, a well-known regulator of EMT3, 4 as well as several other cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-8 and IL-6.5, 6, 7, 8
Oncostatin M (OSM), a member of the IL-6 family, was originally identified as an inhibitor of melanoma cell growth in vitro.9 It was also shown to suppress proliferation but induce mesenchymal-like differentiation of several breast cancer cell lines in vitro.10, 11 Another study reported that activated peripheral blood mononuclear cell-conditioned medium, which contains high levels of OSM, promoted the detachment and invasive capacity of breast cancer cells.12 Granulocyte-macrophage colony-stimulating factor produced by breast cancer cells or cell–cell contact can trigger the release of OSM from neutrophils, implicating that this cytokine is present in the tumor microenvironment and may have a potential role in promoting tumor invasion and metastasis.13
Multiple lines of evidence suggest that inflammatory mediators may interfere with the EMT activators, such as Snail, ZEB and TWIST family members.6, 14, 15 Interplay of these transcription factors represses the expression of E-cadherin, leading to the disruption of intercellular junctions, loss of cellular polarity and acquisition of migratory and invasive phenotypes.16 MicroRNAs (miRNAs) are emerging as powerful regulators of EMT. It is recognized that the expression of miRNAs can be affected by inflammatory cytokine-mediated EMT.17 The miR-200 family has been known as a silencer of ZEB1 and ZEB2. The critical role of the TGF-β/ZEB1/miR-200 signaling network in regulating EMT has been indicated by several recent studies.14, 18 The positive feedback loop of Lin-28–let-7–IL-6 is another important signaling network, which is activated through Src-mediated TNF-α/nuclear factor (NF)-κB signaling pathway.19 These findings highlight the complexity of the signaling networks orchestrated by transcription factors and miRNAs during EMT. However, how these EMT regulatory networks are controlled remains unclear.
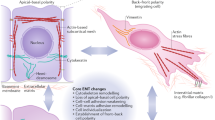
In this study, we demonstrate that OSM is a novel inducer of EMT both in vitro and in vivo. Our data indicate that Lin-28–let-7–HMGA2 (high-mobility group A protein 2) and miR-200–ZEB1 signaling circuits, controlled by OSM-induced signal transducer and activator of transcription factor 3 (Stat3) activation, are responsible for initiating and maintaining the EMT genetic program, revealing a novel mechanism of inflammatory cytokine-mediated phenotypic reprogramming in human breast cancer.
Results
OSM is expressed in invasive breast carcinoma and induces EMT in breast cancer cells
We analyzed the expression of OSM in benign or malignant human breast tissues by immunohistochemistry. The expression of OSM was almost absent in the majority of benign breast tissues (9/10) or pericancerous tissues (Figure 1a). However, 74% (20/27) of cases of invasive breast carcinomas were OSM-positive. Strong staining of OSM was observed in both tumor cells and inflammatory or stromal cells, indicating that OSM was expressed in an autocrine/paracrine fashion in invasive breast carcinoma. We utilized a breast cancer tissue array (BR1503) to analyze the association of OSM expression with clinicopathological characteristics. The above-mentioned results were strongly confirmed as OSM was predominantly expressed in both tumor and stromal cells in malignant tissues (∼80%) but rarely in normal or benign tissues (Supplementary Table S1). We found that OSM expression seems to be slightly correlated with ErbB2 (P=0.053) in breast cancer (Supplementary Table S1).

OSM is expressed in an autocrine/paracrine fashion in invasive breast carcinoma. (a) The expression of OSM in human breast cancer tissue samples were examined by immunohistochemistry with rabbit polyclonal antibody against OSM. The representative images from the area adjacent to the tumor tissue (left panel) and tumor tissue (right panel) in the same tissue section are shown. (b) The expression levels of OSM and CD68 in breast cancer tissues were detected in serial sections by immunohistochemistry with the anti-OSM antibody and mouse monoclonal antibody against CD68. Bar=25 μM. (c) THP-1 cells were treated with 320 nM PMA (phorbol 12-myristate 13-acetate) for 48 h. For generation of M1- and M2-polarized macrophages, THP-1 cells were treated with 320 nM PMA for 6 h and then cultured with PMA plus 20 ng/ml IL-4 and 20 ng/ml IL-13 (for M2 polarization) or plus 100 ng/ml lipopolysaccharide and 20 ng/ml interferon-γ (for M1 polarization) for another 42 h. The expression of OSM was analyzed by western blot. (d) The expression of OSM and vimentin in human breast cancer cell lines was analyzed by western blot. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Notably, in the central region of some tumor tissues, the expression of OSM was not detectable, whereas positive staining was distinctively detected at the invasive front (Supplementary Figure S1). It has been shown that tumor-associated macrophages are frequently localized at the invasive front of more advanced malignancies.20 By immunohistochemical labeling in serial sections for OSM and CD68, a macrophage marker, many OSM-positive cells were identified as macrophages (Figure 1b). Interestingly, a large amount of OSM-expressing inflammatory cells were frequently observed around the tumor sinusoids or inflammatory lesions, implying that OSM may provide an autocrine stimulus for the migration and infiltration of inflammatory cells (Supplementary Figures S2A and B). In addition, by western blot and enzyme-linked immunosorbent assay analysis of the OSM expression and secretion in M1- or M2-polarized human monocytic THP-1 cells, we demonstrated that M2-polarized macrophages produced significantly higher level of OSM than M1-polarized macrophages (Figure 1c, Supplementary Figures S2C and D). We also examined OSM expression in a panel of human breast cancer cell lines. Figure 1d shows that the level of OSM was low or undetectable in non-invasive or low invasive T47D, MCF-7, BT474, MDA-453 and SKBR3 cells, whereas pronounced expression of OSM was detected in highly invasive MDA-231 and MDA-435s cells. Additionally, the level of a mesenchymal marker, vimentin, was also high in MDA-231 and MDA-435s cells, but seldom detectable in the other cell lines tested. These data suggest an important role of an autocrine/paracrine secretion of OSM in the progression of breast cancer.
It has been demonstrated that OSM can alter mammary cell morphology from a normal epithelial phenotype to a mesenchymal-like phenotype,11 suggesting that OSM may be a novel inducer of EMT. We explored the effects of OSM on EMT in two luminal cell lines, MCF-7 and T47D, which represent a non-advanced stage of breast cancer malignancy. As displayed in Figure 2a, the expression levels of the mesenchymal markers, fibronectin and vimentin, and the transcription repressors, ZEB1 and Snail of E-cadherin, were dramatically increased in MCF-7 cells after 6 days of OSM treatment, while the expression level of epithelial marker E-cadherin was progressively reduced. Similar results were also observed in T47D cells (Supplementary Figure S3A). Without OSM stimulation, E-cadherin was predominantly localized at the cytoplasmic membranes of MCF-7 cells, and no fibronectin expression was detected. After OSM stimulation for 5 days, the E-cadherin expression was greatly decreased, whereas a prominent perinuclear aggregation of fibronectin was visualized, concomitant with changes in cell shape (Figure 2b and Supplementary Figure S3B). This EMT-like process was accompanied with the induction of a variety of proinvasive and prometastatic genes, including matrix metalloproteinases (MMPs), COX-2, vascular endothelial growth factor and the chemokine receptor CXCR4 (Supplementary Figure S3C), suggesting that these cells acquired malignant traits. By in vitro wound-healing and Matrigel invasion assays, we demonstrated that OSM treatment strongly induces invasive and migratory properties in MCF-7 cells. As exhibited in Figure 2c, a more rapid and complete wound closure occurred in the presence of OSM than in the absence of OSM. Moreover, OSM quickly induced the invasive activities of the cells as early as 12 h after treatment, and the activities were continuously increased until day 3 (Figures 2d and e). Based on the alterations of the phenotype and biological behaviors, we conclude that OSM induces EMT in breast cancer cells and enhances cell migration and invasion.

OSM induces EMT in breast cancer cells and enhances cell migration and invasion. (a) MCF-7 cells were treated with 10 ng/ml OSM and the expression levels of E-cadherin, fibronectin, vimentin, ZEB1 and Snail were analyzed by western blot at the indicated time points. (b) MCF-7 cells were treated with phosphate-buffered saline or 10 ng/ml of OSM for 5 days. The expression levels of E-cadherin and fibronectin were examined by immunofluorescence/confocal microscopy. Bar=50 μM. (c) The migratory activities were determined by the wound-healing assays at the indicated days after OSM stimulation. (d, e) The invasive activities of MCF-7 cells were determined by Matrigel invasion assays. The invading cells were stained (d) and quantified (e). Error bars represent s.e. (N⩾3). NC, mimics negative control; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Downregulation of let-7 and miR-200 levels initiates OSM-induced EMT phenotypes
Two evolutionary conserved miRNA families let-7 and miR-200 have been identified as powerful regulators of EMT.19, 21, 22 Let-7 targets a variety of molecules involved in differentiation and early embryogenesis, and miR-200 directly regulates E-cadherin transcriptional repressors, ZEB1 and ZEB2.22 To determine whether let-7 and miR-200 are engaged in OSM-induced EMT, we analyzed their temporal expression profiles in MCF-7 cells by real-time reverse transcriptase (RT)–PCR. The data showed that the four members of the let-7 family (let-7b, let-7d, let-7e and let-7g) were rapidly repressed after OSM treatment for 6 h, with an approximately 2–4-fold reduction at the 12 h time point. However, at the 72 h time point, the expression of let-7b/d/e/g was re-elevated (Figure 3a). Notably, downregulation of the miR-200 family members, miR-200b and miR-200c, occurred later at 12–24 h but retained at the 72 h time point (Figure 3b). The expression pattern of miR-200b/c was coincident with the elevation of the ZEB1 level. These data revealed that downregulation of let-7 and miR-200, in response to OSM stimulation, exhibited different temporal patterns.

Downregulation of let-7 and miR-200 levels initiates and maintains OSM-induced EMT phenotype. (a, b) MCF-7 cells were treated with 10 ng/ml of OSM and the expression levels of the let-7 (a) and miR-200 (b) families were analyzed by real-time RT–PCR at the indicated time points. (c) MCF-7 cells were transfected with let-7b/d/g and miR-200b/c. After transfection for 24 h, the cells were treated with 10 ng/ml of OSM for 5 days. The expression of E-cadherin, fibronectin, ZEB1 and Snail was analyzed by western blot. (d) MDA-231 cells were transfected with let-7b/d/g and miR-200b/c for 48 h. The expression levels of E-cadherin, fibronectin, vimentin and ZEB1 were analyzed by western blot. (e, f) MCF-7 and MDA-231 cells were transfected with let-7b/d/g and miR-200b/c. The transfected MCF-7 cells were treated with 10 ng/ml of OSM for 5 days. The invasive activities of the cells were determined by Matrigel invasion assays after 10 h (MDA-231 cells) or 48 h (MCF-7 cells) of culture. The invading cells were stained (e) and quantified (f). (g) MCF-7 cells were co-transfected with let-7 inhibitors and miR-200 mimics for 24 h. Then the cells were treated with OSM. The expression levels of E-cadherin, fibronectin and ZEB1 were analyzed by western blot after OSM induction for 4 days. Error bars indicate s.e. (N⩾3). NC, nonspecific miRNA mimics; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
We transfected let-7b/d/g and miR-200b/c into MCF-7 cells and demonstrated that forced expression of these exogenous miRNAs dramatically restored the expression of E-cadherin and downregulated the levels of ZEB1, Snail and fibronectin (Figure 3c). The reversal of OSM-induced phenotypes by let-7 and miR-200 overexpression was confirmed by immunostaining (Supplementary Figure S4). Similar data were obtained in MDA-231 cells that secrete OSM in an autocrine manner (Figure 3d). Moreover, the migratory and invasive capabilities of MDA-231 and OSM-treated MCF-7 cells were effectively abolished by ectopic expression of let-7 and miR-200 (Figures 3e and f). To further verify the role of transient let-7 repression in OSM-induced EMT, we co-transfected MCF-7 cells with let-7 inhibitors and miR-200 mimics (Figure 3g). We observed that let-7 inhibitors apparently potentiated the effect of OSM on EMT. Interestingly, let-7 inhibitors markedly antagonized the ZEB1 suppression and phenotype reversal by miR-200 mimics. Taken together, these data suggest that downregulation of let-7 and miR-200 by OSM triggers the phenotypic transition in breast cancer cells and that let-7 has a critical role, independent of miR-200, in early stage of OSM-induced EMT.
Stat3 activation mediates OSM-induced repression of let-7 and miR-200
Previous studies have suggested that aberrant activation of signaling pathways by inflammatory cytokines, such as TGF-β, TNF-α and IL-6, induces EMT and malignant transformation.5, 6, 14 Classical transcription factors, Stat3, NF-κB, activator protein 1 (AP-1) and Smad, are central signaling hubs in inflammation-mediated tumor promotion and metastasis.19, 23, 24 To determine whether these classical transcription factors are involved in OSM-induced EMT, we transfected the luciferase reporter vectors containing the consensus-binding sequences for Stat3, NF-κB, AP-1 and Smad into MCF-7 cells and monitored the transactivation activities of these transcription factors. Luciferase reporter assays showed that OSM significantly induced the activities of AP-1 and Stat3 but not NF-κB and Smad (Supplementary Figure S5A). Notably, the activation of Stat3 and c-Jun started within 1 h after addition of OSM and lasted 6 days (Figure 4a), indicating that OSM stimulation caused a rapid and persistent activation of Stat3 and AP-1. OSM mediates signal transduction mainly through the mitogen-activated protein kinase (MAPK) or the Janus-activated kinase (Jak)/Stat pathways.25 Treatment of the cells with the inhibitors of Jak2/Stat3 (WP1066), MEK1/2 (PD98059) and JNK (SP600125) significantly suppressed the activation of Stat3, abrogated OSM-induced expression of fibronectin and ZEB1 and increased the expression of E-cadherin (Supplementary Figure S5B). These data implicate that OSM-induced EMT is primarily mediated by MAPK/ERK (extracellular signal–regulated kinase), JNK and Jak2 signaling pathways, which all converge on Stat3 activation. To confirm the critical role of Stat3 in OSM-induced EMT, we established a Stat3-deficient MCF-7 cell line MCF-7/Stat3 sh (Figure 4b). Knock down of Stat3 remarkably inhibited OSM-induced upregulation of fibronectin and ZEB1 and restored the expression of E-cadherin (Figure 4c). Similar data were obtained by knock down of Stat3 in MDA-231 cells (Figure 4d). Consistent with the phenotypic alterations, the invasive potential of the cells was considerably attenuated in both MDA-231 and OSM-induced MCF-7 cells (Figures 4e and f). These data indicate that OSM elicits EMT through activating Stat3.

Stat3 activation mediates OSM-induced repression of let-7 and miR-200. (a) MCF-7 cells were treated with 10 ng/ml of OSM and then p-Stat3, p-c-Jun and c-Fos were analyzed by western blot at the indicated time points. (b) MCF-7 cells were stably transfected with the plasmid expressing Stat3-specific shRNA (MCF-7/Stat3 sh). The expression of Stat3 was determined by western blot. (c) The transfected cells were treated with 10 ng/ml of OSM for 4 days. The expression levels of E-cadherin, fibronectin, ZEB1 and p-Stat3 were analyzed by western blot. (d) MDA-231 cells were stably transfected with the plasmid expressing Stat3 shRNA (MDA-231/Stat3 sh). The expression levels of E-cadherin, fibronectin, ZEB1 and Stat3 were analyzed by western blot. (e, f) MCF-7/Stat3 sh cells were treated with 10 ng/ml of OSM for 5 days. The invasive activities of the cells were determined by Matrigel invasion assays after 10 h (MDA-231/Stat3 sh) or 48 h (MCF-7/Stat3 sh) of culture. The invading cells were stained (e) and quantified (f). (g) MCF-7 cells were transfected with the plasmid expressing Stat3C and the expression of let-7b/d/e/g and miR-200b/c was analyzed by real-time RT-PCR after transfection for 48 h. (h, i) The expression levels of let-7e and miR-200c in MCF-7/Stat3 sh cells were analyzed by real-time RT-PCR at the indicated time points after OSM induction. (j) MCF-7 cells were cotransfected with the plasmids containing HMGA2 5′ and 3′ UTRs, ZEB1 3′ UTR and Stat3C. Luciferase activities were measured after transfection for 36 h. Error bars indicate s.e. (N⩾3). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Given that the activation of Stat3 and downregulaion of let-7 and miR-200 were involved in OSM-triggered EMT, we speculated that Stat3 activation may associate with downregulation of the let-7 and miR-200 expression. In agreement with this idea, transfection of Stat3C, a constitutively active Stat3 mutant into MCF-7 cells caused a remarkable reduction of let-7b/d/e/g and miR-200b/c (Figure 4g). Conversely, Knock down of Stat3 thoroughly prevented OSM-induced downregulation of let-7e, while the restoration of miR-200c occurred somewhat later after 24 and 72 h (Figures 4h and i). Moreover, the levels of let-7e and miR-200c were also markedly upregulated by knockdown Stat3 in MDA-231 cells (Supplementary Figure S6A). HMGA2 was recently identified as a target gene of let-7, and its 3′ untranslated region (UTR) harbors seven conserved sites complementary to let-7.26 miR-200 is known to have the strongest inhibitory effect on the expression of ZEB1, which carries five putative miR-200b/c matches in its 3′ UTR.21 To test whether the expression of let-7 and miR-200 is regulated by Stat3, we constructed luciferase reporter plasmids bearing ZEB1 or HMGA2 3′ UTRs. The repressive effects of let-7 and miR-200 on HMGA2 and ZEB1 3′ UTRs were confirmed by luciferase assays (Supplementary Figures S6B and C). Figure 4j showed that Stat3C significantly enhanced the activities of HMGA2 and ZEB1 3′ UTRs, but failed to activate HMGA2 5′ UTR. Transfection with the Stat3C expression plasmid significantly increased the transcription of HMGA2 gene and led to the upregulation of HMGA2 protein level in a concentration-dependent manner (Supplementary Figures S7A and B). These data indicate that Stat3 upregulates the expression of HMGA2 and ZEB1 at the post-transcriptional level by suppressing let-7 and miR-200.
HMGA2 acts as a master switch of OSM-induced-EMT
Above data revealed that OSM induced a transient downregulation of let-7 and sustained repression of miR-200. A direct mechanism linking miR-200, ZEB1, E-cadherin and EMT can be easily understood in the context of the current literatures.27, 28 However, the significance of a transient repression of the let-7 family in OSM-induced EMT is difficult to interpret. Let-7 is a well-recognized tumor suppressor and targets a number of oncogenes, such as Ras, c-myc and HMGA2.22 In response to OSM induction, the let-7 expression rapidly declined, reaching a minimum around 12 h followed by an abrupt reversal at 72 h, whereas the expression of c-Myc, Ras and HMGA2 swiftly elevated, approaching a maximum, coincidentally, around 12 h followed by a recovery back to its baseline level or even disappearance of the expression (Figure 5a). Ectopic expression of let-7 fully suppressed the expression of Ras, c-myc and HMGA2 in MDA-231 and OSM-treated MCF-7 cells (Figures 5b and c). As HMGA2 is known to control the expression of a diverse set of transcription factors involved in regulation of E-cadherin transcription,29, 30 we considered the possibility that HMGA2 may act as a powerful regulator in OSM-induced EMT. As shown in Figure 5d, knock down of HMGA2 in mesenchymal-like MDA-231 cells potently upregulated the E-cadherin expression and downregulated the fibronectin expression in a dose-dependent manner. When OSM-induced HMGA2 in MCF-7 cells was repressed by HMGA2 siRNA, the EMT phenotype was dramatically reversed (Figures 5e and f), and the expression of miR-200c was also restored (Figure 5g). Furthermore, the invasive capabilities were significantly impaired in both MDA-231 and OSM-treated MCF-7 cells (Figures 5h and i), demonstrating that the expression of HMGA2 in breast cancer cells caused a switch from an epithelial to a mesenchymal phenotype. As mentioned previously, transient repression of let-7 was controlled by activated Stat3. Figure 5j illustrates that when Stat3 was knocked down in MCF-7 cells, OSM-induced HMGA2 expression was fully inhibited, confirming that the let-7–HMGA2 axis is dominated by Stat3 activation. These data indicate that transient induction of HMGA2 and repression of let-7 may initiate an intermediate transitional stage of EMT, implicating a novel mechanism underlying the initiation and maintenance of EMT.

HMGA2 acts as a master switch of EMT. (a) MCF-7 cells were treated with 10 ng/ml of OSM and the expression levels of c-Myc, Ras and HMGA2 were determined by western blot at the indicated time points. (b) MDA-231 cells were transfected with let-7b/d/g and the expression levels of c-Myc, Ras and HMGA2 were analyzed by western blot. (c) MCF-7 cells were transfected with let-7b/d/g. After transfection for 24 h, the cells were exposed to 10 ng/ml of OSM. The expression levels of c-Myc, Ras and HMGA2 were analyzed by western blot at the indicated time points. (d) MDA-231 cells were transfected with 50 or 100 nM HMGA2-specific siRNA. After transfection for 48 h, the expression levels of E-cadherin, fibronectin and HMGA2 were analyzed by western blot. (e) MCF-7 cells were transfected with HMGA2 siRNA. After transfection for 48 h, the cells were treated with 10 ng/ml of OSM. The expression of HMGA2 was analyzed by western blot at the indicated time points. (f, g) MCF-7 cells were transfected with HMGA2 siRNA. After transfection for 24 h, the cells were treated with 10 ng/ml of OSM for 4 days. The expression levels of fibronectin, E-cadherin and ZEB1 were determined by western blot (f) and miR-200c by real-time RT–PCR (g). (h, i) MCF-7 and MDA-231 cells were transfected with HMGA2 siRNA. The transfected MCF-7 cells were treated with 10 ng/ml of OSM for 4 days. The invasive activities were determined by Matrigel invasion assays after 10 h (MDA-231 cells) or 48 h (MCF-7 cells) of culture. The invading cells were stained (h) and quantified (i). (j) MCF-7/Stat3 sh cells were exposed to 10 ng/ml of OSM. The expression of HMGA2 and phosphorylation of Stat3 were analyzed by western blot at the indicated time points. Error bars indicate s.e. (N⩾3). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Lin-28B transactivaion by Stat3 contributes to transient let-7 downregulation
It has been reported that the Lin-28 and Lin-28B RNA-binding proteins block let-7 processing into mature miRNAs by directly binding to the terminal loops of the precursors of the let-7 family.31 Given that Stat3 activation led to decreased expression of let-7, we speculated that OSM stimulation may modulate the expression level of let-7 via upregulation of Lin-28B through Stat3. To test this hypothesis, we examined the alteration of the Lin-28B expression in response to OSM induction. At the same time, we assessed the levels of primary let-7e and mature let-7e by real-time RT–PCR. Our data demonstrated that the Lin-28B expression was dramatically increased after OSM treatment, starting to rise at 6 h and peaking at 12–24 h after treatment, coincident with the reduction of mature let-7e level (Figure 6a). However, the level of pri-let-7e was not changed, reflecting Lin-28-mediated inhibition of let-7 processing. We then transfected Stat3C into MCF-7 cells and detected the expression of Lin-28B. As expected, the Lin-28B expression was strikingly enhanced (Figure 6b). Conversely, the effect of OSM stimulation on Lin-28B expression was attenuated in MCF-7/Stat3 sh cells (Figure 6c).

Transcriptional activation of Lin-28B by Stat3 contributes to transient let-7 downregulation. (a) MCF-7 cells were exposed to 10 ng/ml of OSM. The expression levels of primary let-7e (pri-let-7e) and mature let-7e (mat-let-7e) were analyzed by real-time RT–PCR and Lin-28B by western blot at the indicated time points. (b) MCF-7 cells were transfected with the Stat3C expression plasmid. The expression of Lin-28B was analyzed by western blot. (c) MCF-7/Stat3 sh cells were exposed to 10 ng/ml of OSM. The expression of Lin-28B and phophorylation of Stat3 were analyzed by western blot at the indicated time points. (d) Two Stat3 sites were identified at the upstream of the −2405 and −369 bp of the Lin-28 5′ UTR. (e) MCF-7 cells were transfected with the Flag-Stat3C expression plasmid. Binding of Stat3 to the Lin-28 5′ UTR was determined by ChIP assays using the anti-Flag antibody. (f) MCF-7 cells were exposed to 10 ng/ml of OSM and binding of endogenous Stat3 to the Lin-28 5′ UTR was determined by ChIP assays using the anti-Stat3 antibody at the indicated time points. (g) MCF-7 cells were transfected with Lin-28 siRNA. After transfection for 24 h, the cells were treated with OSM and the expression levels of E-cadherin, fibronectin, ZEB1 and Lin-28B were analyzed by western blot. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Scrutinizing the Lin-28 promoter sequences for putative Stat3-binding elements, we identified two consensus Stat3 sites at the upstream of the −2405 and −369 bp of the Lin-28 5′ UTR (Figure 6d). To determine whether Stat3 physically interacts with the Lin-28 promoter, we transfected MCF-7 cells with the Flag-Stat3C expression plasmid and performed chromatin immunoprecipitation (ChIP) assays by using an antibody against Flag. The data demonstrated that Stat3 bound to both potential Stat3 sites within the Lin-28 promoter (Figure 6e). In accord with the kinetics of Lin-28 expression, strong binding of endogenous Stat3 was observed at 12 h after OSM induction, but not in the absence of OSM (Figure 6f), as determined by ChIP assays using the anti-Stat3 antibody. Further knock-down of Lin-28B dramatically reversed the repressive effect of OSM on the expression of E-cadherin and inhibited OSM-induced upregulation of fibronectin and ZEB1 (Figure 6g). The negatively correlated expression patterns between Lin-28B/let-7 and let-7/HMGA2 indicate that Stat3 activates the transcription of Lin-28B, leading to the downregulation of let-7 and subsequent upregulation of HMGA2. These data reveal a mechanism behind the phenotypic transition of breast cancer cells after OSM induction.
OSM induces EMT in breast cancer xenografts in nude mice
To investigate the effects of OSM on EMT and breast cancer metastasis in vivo, we inoculated MCF-7/GFP (green fluorescent protein) cells into the inguinal fat pads of NOD/SCID mice. After the tumors were palpable, OSM was injected into the tumors at a dosage of 100 ng/kg weekly for six consecutive cycles. After 2 months, the animals were killed and their tumors dissected. Figures 7a and b showed that OSM treatment slightly accelerated tumor growth in mice, though the increase in tumor size was not statistically significant (P=0.53). However, in agreement with our in vitro data, OSM stimulation greatly promoted the invasive capacities of MCF-7/GFP cells and caused spontaneous lung metastasis in six out of eight mice, whereas no metastasis was observed in the control group. Metastatic colonization was observed by microscopic inspection of tissue sections and identified by immunohistochemistry using the anti-GFP antibody (Figure 7c), demonstrating that OSM stimulation confers the invasive potential to breast cancer cells in vivo. In the tumor tissues injected with OSM, Stat3 was remarkably phosphorylated, concomitant with the elevation of fibronectin and ZEB1 and reduction of E-cadherin. The expression of proinvasive and prometastatic molecules (MMPs, COX-2 and CXCR4) was dramatically induced (Figure 7d). We examined the expression of let-7e and miR-200c in the tumor tissues. Interestingly, we found that there was no striking difference in let-7 expression levels between control and OSM-injected tumors. However, the expression of miR-200 was definitely reduced in OSM-injected tumor tissues (Figures 7e and f), confirming that the downregulation of miR-200 expression under OSM stimulation was sustained in vivo. These results strongly suggest that OSM acts in vivo to initiate EMT, promoting the invasiveness and metastases of breast cancer.

OSM promotes lung metastasis of breast cancer xenografts. (a) In all, 2.5 × 106 MCF-7/GFP cells were inoculated into the right inguinal fat pad of each BALB/c female nude mice (4–5 weeks old). When the xenografts were palpable, 20 ng OSM (n=8) or phosphate-buffered saline (PBS; n=6) was injected into the tumors once a week for consecutive 6 weeks. The mice were killed after 60 days. Fluorescent images of the mice were acquired by a NightOwl LB 981 Molecular Light Imager (Berthold Technologies, Bad wildbad, Germany). (b) The tumors were dissected and weighed. (c) The tumor and lung tissues were formalin-fixed. The paraffin-embedded sections were subjected to hematoxylin and eosin staining or probed with the anti-GFP antibody. (d) The tumor tissue samples were lyzed and the expression levels of MMP-2, MMP-7, MMP-9, COX-2, CXCR4, E-cadherin, vimentin, ZEB1, Stat3 and p-Stat3 were analyzed by western blot. (e, f) The expression levels of let-7e and miR-200c in the tumor tissues injected with PBS or OSM were evaluated by real-time RT–PCR. (g) Diagram of the mechanism of Stat3-coordinated Lin-28–let-7–HMGA2 and miR-200–ZEB1 circuits initiate and maintain OSM-driven EMT.
Discussion
Inflammatory cytokines in tumor microenvironment can promote the migratory and invasive abilities of tumor cells through the induction of EMT, a key event triggering cancer cell dissemination and metastasis.32, 33, 34 Several studies suggest that OSM induces EMT in breast cancer cells. High expression of the OSM receptor in cervical squamous cell carcinoma cells has been associated with increased cell motility and invasiveness, implicating a potential role of OSM in tumor progression.35 However, the effect of OSM on EMT in human cancer cells in vivo has not been explored.36 In this study, we show that the autocrine/paracrine signaling of OSM by tumor and inflammatory cells, particularly macrophages, closely correlated with invasive characteristics of human breast cancers, that OSM stimulation was sufficient to induce EMT not only in vitro but also in vivo and that OSM promoted spontaneous lung metastases in MCF-7 breast cancer xenograft mouse model.
Inflammatory cytokine-triggered EMT can be a consequence of Stat3 activation. The transcriptional suppressors of E-cadherin, Twist, Snail and ZEB1 have been identified as direct transcriptional targets of Stat3.5, 37, 38, 39 It has also recently been shown that Stat3 activation through Ras signaling pathway was required for EMT in salivary epithelial cells,40 and ectopic expression of Stat3C decreased the E-cadherin level in prostate epithelial cells.41 Our data indicate that persistent activation of Stat3 significantly perturbed the expression of key miRNAs let-7 and miR-200 families, resulting in comprehensive alterations of the transcription factors and oncoproteins targeted by let-7 or miR-200 in breast cancer cells. Blocking of Stat3 signaling pathway or knock down of Stat3 effectively reversed the mesenchymal phenotype and restored the expression of let-7 and miR-200 families in OSM-treated MCF-7 cells. These data indicate an obligatory role for Stat3 in governing the network of transcription factors and miRNAs involved in EMT.
Concomitant with transient reduction of the let-7 level after exposure to OSM, the expression of HMGA2 was instantly upregulated, accompanied by an increase in the levels of mesenchymal markers. The expression of the let-7 family members in human is barely detectable in embryonic stages but upregulated at the end of embryonic development.42, 43 Conversely, HMGA2 as an early embryonic gene is highly expressed in undifferentiated proliferating cells during embryogenesis and becomes silent in adult tissues.22, 44 HMGA2 codes for a small, nonhistone chromatin architectural factor that can modulate transcription by inducing DNA conformational changes.30 It has been demonstrated that HMGA2 cooperates with Smad3 to bind to the Snail promoter and promotes the transcription of Snail, participating in TGF-β-induced EMT in mammary epithelial cells.29 Our data demonstrate that inhibition of HMGA2 by the specific siRNA markedly impeded OSM-driven EMT, interrupted the miR-200–ZEB1 negative feedback loop and reversed OSM-induced invasive phenotype of breast cancer cells.
A recent study reported that IL6-mediated inhibition of miR-200c, which is responsible for suppression of inflammation and cell transformation, directs constitutive activation of inflammatory signaling circuit and drives transformation and tumorigenesis.45 It is proposed that inflammatory cells can ignite transient signal activation. Our current study revealed a similar dynamic mechanism, indicating that transient let-7 repression and HMGA2 induction initiate OSM-induced EMT, while sustained reduction of miR-200 may be necessary for maintaining a mesenchymal phenotype. These data suggest that HMGA2 acts as a master switch for epigenetic transition in human breast cancer cells. The precise molecular mechanisms remain to be explicated.
The RNA-binding protein Lin-28 was recently demonstrated to negatively regulate let-7 biogenesis.31 We found that the transient increase in the Lin-28 paralleled the transient decrease in mature let-7 level. The current study, for the first time, reveals that Stat3 regulates the transcriptional activation of Lin-28B and Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain OSM-driven EMT (Figure 7g). Taken together, these findings indicate that Stat3 signaling is of central importance in controlling multiple effectors in inflammatory cytokine-mediated EMT.
Accumulating data suggest that EMT is mechanistically associated with cancer stem cells.2, 46 It has been proposed that autocrine signaling of inflammatory cytokines may regulate the disparate processes of self-renewal and maintenance of breast cancer stem cells through Stat3-mediated signaling pathways.47, 48 Forced expression of let-7 in stem cells has been shown to induce differentiation and downregulation of the let-7 family members connected with self-renewal of cancer stem cells.49 Lin-28 along with OCT4, SOX2 and NANOG, which are core pluripotent transcription factors, can reprogram somatic murine and human cells into pluripotent stem cells.50 HMGA2 was also demonstrated to be involved in differentiation of embryonic stem cells.51 Several recent studies implicated that cytokine-dependent activation of Stat3 may drive the self-renewal of embryonic stem cells and that ectopic Stat3 activities promote the maintenance of pluripotency.47, 48 Based on these findings and data in this study, we propose a dynamic switch mechanism, by which the Lin-28-let-7-HMGA2 signaling circuit in control of Stat3-mediated inflammatory pathways may regulate self-renewal and differentiation, in cancer stem cells.
In summary, we have identified a central role of OSM-induced Stat3 signaling pathway in controlling the transcriptional network regulating EMT. Prolonged activation of Stat3 leads to the alterations in the expression of the let-7 and miR-200 family members, contributes to the acquisition of the mesenchymal phenotype and invasive capability and promotes the progression of breast cancer.
Materials and methods
Constructs and transfection
The 3′UTRs of HMGA2 and ZEB1 were PCR amplified from the genomic DNA of MDA-231 cells and inserted into the Xba I/EcoR I or Pst I/EcoR I sites downstream of a firefly luciferase gene in a modified pGL3-control reporter vector, respectively, as described earlier.52 A 1608-bp fragment of human HMGA2 promoter was amplified and cloned into the pGL3-basic plasmid (Promega, San Luis Obispo, CA, USA) through the Bgl II and Hind III sites. Primer sequences are shown in Supplementary Table S2. The plasmids pRc/CMV-Stat3C-Flag and luciferase reporter vectors containing the binding sequences for Stat3, NF-κB, AP-1 and Smad were kindly provided by Prof Xuemin Zhang (National Center of Biomedical Analysis, Beijing, China).
Cells were seeded at 1 × 105 cells per well into 12-well plates and transfected with miRNAs at a final concentration of 60 nM (30 nM of each miR-200b and miR-200c or 20 nM of each let-7b, let-7d and let-7g) using Lipofectamine RNAiMAX (13778, Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. After transfection for 24 h, the cells were stimulated with 10 ng/ml of OSM (HPA029814, Sigma, St Louis, MO, USA). The transfection with miRNAs was repeated once every 2–3 days for up to 5 days. For knockdown of HMGA2, Stat3 and Lin-28, 50 nM specific siRNAs (Ribobio Co. Ltd., Guangzhou, China) or the control siRNA was transfected into MDA-231 or MCF-7 cells using Lipofectamine 2000 (Invitrogen).
Western blot analysis
Western blotting was performed as described previously.53 Cell extracts were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot analysis with the antibodies against E-cadherin (sc-7870, Santa Cruz, Santa Cruz, CA, USA), fibronectin (sc-18825, Santa Cruz), Snai1 (sc-28199, Santa Cruz), c-Fos (sc-52, Santa Cruz), HMGA2 (sc-30223, Santa Cruz), c-Myc (sc-40, Santa Cruz), Stat3 (sc-482, Santa Cruz), vimentin (3932, Cell Signaling Technology, Beverly, MA, USA), p-Stat3 (9145, Cell Signaling Technology), p-c-Jun (9164, Cell Signaling Technology), Lin-28B (4196, Cell Signaling Technology), Ras (3339, Cell Signaling Technology), ZEB1 (ab23398, Abcam, Cambridge, UK) and OSM. Bands were visualized by the Enhanced Chemiluminescence System (Thermo Scientific, Waltham, MA, USA).
Immunohistochemistry
Breast tissue samples were obtained from Donghua Hospital, Dongguan, Guangdong, China, with informed consent of patients and approval for the experiments from Donghua Hospital. Human breast cancer tissue arrays (Cat. No. BR1503), including 3 normal tissues, 3 fibroadenomas, 2 cystosarcomas phyllodes, 7 intraductal carcinomas and 60 invasive ductal carcinoma, were purchased from US Biomax (Rockville, MD, USA). Immunohistochemistry was carried out as previously described,53 using rabbit polyclonal antibody against OSM (HPA029814, Sigma) and mouse monoclonal antibody against CD68 (ZM-0060, Zhongshan Golden Bridge, Beijing, China). Digital image was collected under an Olympus BX53 microscope (Olympus, Tokyo, Japan).
ChIP assays
The ChIP assays were performed with SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling Technology) according to the manufacturer’s protocol. The antibody against Flag (M2, Sigma) and Stat3 (sc-482X, Santa Cruz) were used to evaluate the binding of the exogenous or endogenous Stat3 to the Lin-28B promoter. Immunoglobulin G was used as the negative control. Precipitated DNA was analyzed by PCR using the specific primers (Table S2).
Animal studies
17β-Estradiol pellets (0.72 mg, 60-day release; SE-121, Innovative Research, Sarasota, FL, USA) were implanted subcutaneously into 4–5 weeks old BALB/c female nude mice. One day after pellet implantation, 2.5 × 106 MCF-7/GFP cells were injected subcutaneously into the right inguinal fat pad of each mouse. When the xenografts were palpable, 20 ng OSM (n=8) or phosphate-buffered saline (n=6) was injected into the tumors once a week for consecutive 6 weeks. The mice were killed after 60 days and the tumors dissected, weighed and divided into two equal parts. One part of each tumor was formalin-fixed, and the lung and liver tissues were harvested from the same mouse. The paraffin-embedded sections were subjected to routine hematoxylin and eosin staining or probed with the anti-GFP antibody (598, MBL, Woburn, MA, USA). Photographs were taken under an Olympus BX53 microscope. The tumor samples were lyzed and subjected to SDS-PAGE and immunoblot analysis with the antibody against E-cadherin, vimentin, ZEB1, COX-2 (sc-1746, Santa Cruz), CXCR4 (SRP00701, SAIER Biotechnology, Tianjin, China), MMP-2 (MS-567-P0, Neomarker, Waltham MA, USA), MMP-7 (sc-80205, Santa Cruz) and MMP-9 (MS-815-P0, Neomarker). The total RNAs were extracted, and the expression levels of let-7e and miR-200c were determined by real-time PCR. All animal studies were performed in accordance with the institutional guidelines, and the experiments were approved by the Institutional Animal Care and Use Committee at the Institute of Basic Medical Sciences.
Statistical analysis
Statistical analyses were performed with SAS statistical software Version 9.2 (SAS Institute Inc., 2008, Cary, NC, USA). For comparisons among the groups in the experiments, an analysis of variance test was utilized. Fisher’s exact test was used to determine the association between OSM expression, clinical staging and ER/PR/erbB2 status in breast cancer tissues. P<0.05 was considered statistically significant.
References
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A . Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009; 30: 1073–1081.
Biddle A, Mackenzie IC . Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev 2012; 31: 285–293.
Yang L . TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev 2010; 29: 263–271.
Zavadil J, Bottinger EP . TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005; 24: 5764–5774.
Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009; 28: 2940–2947.
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP . Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009; 15: 416–428.
Strippoli R, Benedicto I, Foronda M, Perez-Lozano ML, Sanchez-Perales S, Lopez-Cabrera M et al. p38 maintains E-cadherin expression by modulating TAK1-NF-kappa B during epithelial-to-mesenchymal transition. J Cell Sci 2010; 123: 4321–4331.
Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C . IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res 2011; 71: 5296–5306.
Grant SL, Begley CG . The oncostatin M signalling pathway: reversing the neoplastic phenotype? Mol Med Today 1999; 5: 406–412.
Douglas AM, Grant SL, Goss GA, Clouston DR, Sutherland RL, Begley CG . Oncostatin M induces the differentiation of breast cancer cells. Int J Cancer 1998; 75: 64–73.
Jorcyk CL, Holzer RG, Ryan RE . Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine 2006; 33: 323–336.
Nightingale J, Patel S, Suzuki N, Buxton R, Takagi KI, Suzuki J et al. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J Am Soc Nephrol 2004; 15: 21–32.
Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL . Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 2005; 65: 8896–8904.
Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell 2011; 22: 1686–1698.
Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN . Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 2003; 112: 169–180.
Kalluri R, Weinberg RA . The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119: 1420–1428.
Bhaumik D, Patil CK, Campisi J . MicroRNAs: an important player in maintaining a balance between inflammation and tumor suppression. Cell Cycle 2009; 8: 1822.
Brabletz S, Brabletz T . The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep 2010; 11: 670–677.
Iliopoulos D, Hirsch HA, Struhl K . An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009; 139: 693–706.
Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol 2005; 23: 953–964.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601.
Peter ME . Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle 2009; 8: 843–852.
Jarnicki A, Putoczki T, Ernst M . Stat3: linking inflammation to epithelial cancer - more than a "gut" feeling? Cell Div 2010; 5: 14.
Eferl R, Wagner EF . AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003; 3: 859–868.
Gomez-Lechon MJ . Oncostatin M: signal transduction and biological activity. Life Sci 1999; 65: 2019–2030.
Lee YS, Dutta A . The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 2007; 21: 1025–1030.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008; 9: 582–589.
Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR et al. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009; 27: 1712–1721.
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A . HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 2008; 283: 33437–33446.
Tan EJ, Thuault S, Caja L, Carletti T, Heldin CH, Moustakas A . Regulation of transcription factor twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J Biol Chem 2012; 287: 7134–7145.
Newman MA, Thomson JM, Hammond SM . Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 2008; 14: 1539–1549.
Kalluri R . EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 2009; 119: 1417–1419.
Lopez-Novoa JM, Nieto MA . Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 2009; 1: 303–314.
Lin WW, Karin M . A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 2007; 117: 1175–1183.
Winder DM, Chattopadhyay A, Muralidhar B, Bauer J, English WR, Zhang X et al. Overexpression of the oncostatin M receptor in cervical squamous cell carcinoma cells is associated with a pro-angiogenic phenotype and increased cell motility and invasiveness. J Pathol 2011; 225: 448–462.
Pollack V, Sarkozi R, Banki Z, Feifel E, Wehn S, Gstraunthaler G et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: differential role of ERK signaling. Am J Physiol Renal Physiol 2007; 293: F1714–F1726.
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 2008; 283: 14665–14673.
Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC et al. Roles of STAT3 and ZEB1 in E-cadherin downregulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem 2011; 287: 5819–5832.
Zhao S, Venkatasubbarao K, Lazor JW, Sperry J, Jin C, Cao L et al. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor beta-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res 2008; 68: 4221–4228.
Zentner MD, Lin HH, Deng HT, Kim KJ, Shih HM, Ann DK . Requirement for high mobility group protein HMGI-C interaction with STAT3 inhibitor PIAS3 in repression of alpha-subunit of epithelial Na+ channel (alpha-ENaC) transcription by Ras activation in salivary epithelial cells. J Biol Chem 2001; 276: 29805–29814.
Azare J, Leslie K, Al-Ahmadie H, Gerald W, Weinreb PH, Violette SM et al. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol Cell Biol 2007; 27: 4444–4453.
Roush S, Slack FJ . The let-7 family of microRNAs. Trends Cell Biol 2008; 18: 505–516.
Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS et al. Identification of let-7-regulated oncofetal genes. Cancer Res 2008; 68: 2587–2591.
Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996; 13: 2439–2446.
Rokavec M, Wu W, Luo JL . IL6-mediated suppression of miR-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol Cell 2012; 45: 777–789.
Singh A, Settleman J . EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29: 4741–4751.
Daheron L, Opitz SL, Zaehres H, Lensch MW, Andrews PW, Itskovitz-Eldor J et al. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells 2004; 22: 770–778.
Ying QL, Nichols J, Chambers I, Smith A . BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003; 115: 281–292.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007; 131: 1109–1123.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318: 1917–1920.
Pfannkuche K, Summer H, Li O, Hescheler J, Droge P . The high mobility group protein HMGA2: a co-regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev 2009; 5: 224–230.
Cui J, Fu H, Feng J, Zhu J, Tie Y, Xing R et al. The construction of miRNA expression library for human. Prog Biochem Biophys 2007; 34: 389–394.
Shi M, Liu D, Duan H, Qian L, Wang L, Niu L et al. The beta2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res Treat 2011; 125: 351–362.
Acknowledgements
We are grateful to Dr Xuemin Zhang (National Center of Biomedical Analysis, Beijing) for kindly providing us the plasmids pRc/CMV-Stat3C-Flag and luciferase reporter vectors containing the binding sequences for Stat3, NF-κB, AP-1 and Smad. We also thank Dr Xiaofei Zheng (Beijing Institute of Radiation Medicine, Beijing) for the gift of the modified pGL3-control plasmid. This work is supported by National Basic Research Program of China (973 Program, No. 2010CB911904) and National Natural Science Foundation of China (Nos. 30972690, 31271440 and 81272232).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Guo, L., Chen, C., Shi, M. et al. Stat3-coordinated Lin-28–let-7–HMGA2 and miR-200–ZEB1 circuits initiate and maintain oncostatin M-driven epithelial–mesenchymal transition. Oncogene 32, 5272–5282 (2013). https://doi.org/10.1038/onc.2012.573
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.573
Keywords
This article is cited by
-
The Role of lncRNA-miR-26a-mRNA Network in Cancer Progression and Treatment
Biochemical Genetics (2023)
-
Non-coding RNAs regulation of macrophage polarization in cancer
Molecular Cancer (2021)
-
Bioactive recombinant human oncostatin M for NMR-based screening in drug discovery
Scientific Reports (2021)
-
Vitamin D protects glomerular mesangial cells from high glucose-induced injury by repressing JAK/STAT signaling
International Urology and Nephrology (2021)
-
The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer
Breast Cancer Research (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}