
Key Points
-
Alzheimer's disease (AD) accounts for most cases of dementia that are diagnosed after the age of 60. Acetylcholinesterase inhibitors and NMDA (N-methyl-D-aspartate) antagonists provide some relief from the symptoms of AD, but no treatment with a strong disease-modifying effect is currently available. This review discusses the current status of research into disease-modifying approaches, with particular reference to anti-amyloid strategies.
-
The two main disease mechanism-based approaches are based on the involvement of two proteins — amyloid-β (Aβ) and tau — in AD pathology. Aβ is the main constituent of senile plaques — one of the key pathological characteristics of AD. Tau is the main component of neurofibrillary tangles, the other hallmark lesion of AD.
-
It would seem attractive to identify brain-penetrable small molecule drugs that interfere with Aβ–Aβ peptide interactions, and, over the past decade, several different assay formats for the identification of nucleation and deposition inhibitors have been described. However, only a few aggregation inhibitors have moved into clinical testing.
-
Anti-amyloid immunotherapy for AD has received considerable attention following reports that amyloid pathology was reduced in an amyloid precursor protein (APP) transgenic mouse model on vaccination with aggregated amyloid-β42 (Aβ42). Clinical trials were terminated after four early reports of meningoencephalitis, but a post-mortem study in one patient showed evidence of plaque reduction.
-
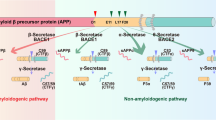
The most direct approach in anti-amyloid therapy is reduction of Aβ42 production. Aβ is generated from APP by the sequential action of β-secretase and γ-secretase. A third protease, α-secretase, can preclude Aβ production by cleaving the peptide in two. This outline points to three strategies to reduce Aβ: inhibition of β-secretase, inhibition of γ-secretase and stimulation of α-secretase.
-
Two key treatment approaches for AD have been driven by retrospective epidemiology: non-steroidal anti-inflammatory drugs and cholesterol-lowering agents. In both cases, the exact target in the disease cascade remains to be elucidated.
-
In the near future, molecules representing several of the strategies outlined in this review will enter clinical trials, so we should find out in the next few years whether the promise of disease modification by any of these strategies is fulfilled. If successful, anti-amyloid drugs would be the first to address the pathogenic mechanism of a CNS disease, and they could become the standard of care in AD.
Abstract
Treating Alzheimer's disease (AD) is the biggest unmet medical need in neurology. Current drugs improve symptoms, but do not have profound disease-modifying effects. Three main classes of disease-modification approaches can be defined: one that is broadly neurotrophic or neuroprotective, one that targets specific aspects of AD pathology, and one that is based on epidemiological observation. This review discusses all three approaches, with particular emphasis on anti-amyloid strategies — currently the most active area of investigation. The approaches that are reviewed include secretase inhibition, amyloid-β aggregation inhibition, immunotherapy and strategies that might indirectly affect the amyloid pathway.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Davis, K. L. & Samuels, S. C. in Pharmacological Management of Neurological and Psychiatric Disorders (eds Enna, S. J. & Coyle, J. T.) 267–316 (McGraw–Hill, New York, 1998).
Doody, R. S. Therapeutic standards in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 13 (Suppl. 2), S20–S26 (1999).
Ferris, S. H. Evaluation of memantine for the treatment of Alzheimer's disease. Expert Opin. Pharmacother. 4, 2305–2313 (2003).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). An updated summary of the amyloid hypothesis.
Walsh, D. M. et al. Naturally secreted oligomers of amyloid protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002).
Gong, Y. et al. Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl Acad. Sci. USA 100, 10417–10422 (2003).
Drews, J. Drug discovery: a historical perspective. Science 287, 1960–1964 (2000).
Cherny, R. A. et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30, 665–676 (2001).
Ritchie, C. W. et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease. Arch. Neurol. 60, 1685–1691 (2003).
Schenk, D. et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 (1999). The first publication on a vaccination approach for AD.
Morgan, D. et al. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985 (2000).
Janus, C. et al. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408, 979–982 (2000).
Hrncic, R. et al. Antibody-mediated resolution of light chain-associated amyloid deposits. Am. J. Pathol. 157, 1239–1246 (2000).
Bard, F. et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Med. 6, 916–919 (2000).
Poduslo, J. F., Curran, G. L., Wengenack, T. M., Malester, B. & Duff, K. Permeability of proteins at the blood-brain barrier in the normal adult mouse and double transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 8, 555–567 (2001).
Sturchler-Pierrat, C. et al. Two amyloid precursor protein transgenic mouse models with Alzheimer's disease like pathology. Proc. Natl Acad. Sci. USA 94, 13287–13292 (1997).
Winkler, D. T. et al. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627 (2001).
Pfeifer, M. et al. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science 298, 1379 (2002).
Bard, F. et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer's disease like neuropathology. Proc. Natl Acad. Sci. USA 100, 2023–2028 (2003).
Monsonego, A. Immunogenic aspects of amyloid β-peptide: implications for pathogenesis and treatment of Alzheimer's disease. Neurobiol. Aging 23 (Suppl.), 112 (2002).
Frenkel, D., Katz, O. & Solomon, B. Immunization against Alzheimer's β-amyloid plaques via EFRH phage administration. Proc. Natl Acad. Sci. USA 97, 11455–11459 (2000).
DeMattos, R. B. et al. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA 98, 8850–8855 (2001).
Dodart, J. C. et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nature Neurosci. 5, 452–457 (2002).
Orgogozo, J. M. et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 vaccination. Neurology 61, 46–54 (2003).
Nicoll, J. A. R. et al. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med. 9, 448–452 (2003). First report to indicate that anti-Aβ immunotherapy reduces plaque pathology in humans.
Ferrer, I., Rovira, M. B., Guerra, M. L. S., Rey, M. J. & Costa-Jussa, F. Neuropathology and pathogenesis of encephalitis following amyloid-β immunization in Alzheimer's disease. Brain Pathol. 14, 11–20 (2004).
Hock, C. et al. Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38, 547–554 (2003).
Nitsch, R. M., Slack, B. E., Wurtman, R. J. & Growdon, J. H. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307 (1992). First demonstration that muscarinic agents can modulate APP processing.
Buxbaum, J. D. et al. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer β/A4 amyloid precursor protein. Proc. Natl Acad. Sci. USA 89, 10075–10078 (1992).
Fisher, A. M1 muscarinic agonists: their potential in treatment and as disease-modifying agents in Alzheimer's disease. Drug Dev. Res. 50, 291–297 (2000).
Hock, C. et al. Treatment with the selective muscarinic m1 agonist talsaclidine decreases cerebrospinal fluid levels of Aβ42 in patients with Alzheimer's disease. Amyloid 10, 1–6 (2003).
Haass, C. et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325 (1992).
Shoji, M. et al. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science 258, 126–129 (1992).
Dovey, H. F. et al. Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 (2001).
Lanz, T. A. et al. The γ-secretase inhibitor N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester reduces Aβ levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque bearing) Tg2576 mice. J. Pharmacol. Exp. Ther. 305, 864–871 (2003).
Siemers, E. R. et al. in 56th Annual Meeting of the American Academy of Neurology abstr. S17.001 (San Francisco, USA, 2004).
DeStrooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active γ-secretase complex. Neuron 38, 9–12 (2003). Recent review of the complex biology of γ-secretase.
Edbauer, D. et al. Reconstitution of γ-secretase activity. Nature Cell Biol. 5, 486–488 (2003).
DeStrooper, B. et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 (1999). Demonstration that Notch signalling depends on γ-secretase activity, affecting the development of γ-secretase inhibitor drugs.
Hadland, B. K. et al. γ-secretase inhibitors repress thymocyte development. Proc. Natl Acad. Sci. USA 98, 7487–7491 (2001).
Doerfler, P., Shearman, M. S. & Perlmutter, R. M. Presenilin-dependent γ-secretase activity modulates thymocyte development. Proc. Natl Acad. Sci. USA 98, 9312–9317 (2001).
Wong, G. T. et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 279, 12876–12882 (2004).
Jarrett, J. T., Berger, E. P. & Lansbury, P. T. Jr. The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 (1993).
Weggen, S. et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 (2001). First publication to show an Aβ42 lowering effect of certain NSAIDs.
Sagi, S., Weggen, S., Eriksen, J. L., Golde, T. E. & Koo, E. H. The non-cyclooxygenase targets of non-steroidal anti-inflammatory drugs, lipoxygenases, peroxisome proliferator-activated receptor, inhibitor of κB kinase, and NFκB, do not reduce amyloid β42 production. J. Biol. Chem. 278, 31825–31830 (2003).
Weggen, S. et al. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid β42 production by direct modulation of γ-secretase activity. J. Biol. Chem. 278, 31831–31837 (2003).
Wolfe, M. S. et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 (1999).
Vassar, R. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999). First identification of β-secretase.
Sinha, S. et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 (1999).
Yan, R. et al. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase specificity. Nature 402, 533–537 (1999).
Hussain, I. et al. Identification of a novel aspartic protease (Asp2) as β-secretase. Mol. Cell. Neurosci. 14, 419–427 (1999).
Saunders, A. J. et al. BACE maps to chromosome 11 and a BACE homolog, BACE2, resides in the obligate Down syndrome region of chromosome 21. Science 286, 1255 (1999).
Citron, M. β-secretase inhibition for the treatment of Alzheimer's disease — promise and challenge. Trends Pharmacol. Sci. 25, 59–112 (2004).
Holsinger, R. M. D., McLean, C. A., Beyreuther, K., Masters, C. L. & Evin, G. Increased expression of the amyloid precursor β-secretase in sporadic Alzheimer's disease. Ann. Neurol. 51, 783–786 (2002).
Fukumoto, H., Cheung, B. S., Hyman, B. T. & Irizarry, M. C. β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389 (2002).
Yang, L. B. et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Med. 9, 3–4 (2003).
Li, R. et al. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc. Natl Acad. Sci. USA 101, 3632–3637 (2004).
Zambrowicz, B. P. & Sands, A. T. Knockouts model the 100 best-selling drugs — will they model the next 100? Nature Rev. Drug Discov. 2, 38–51 (2003).
Luo, Y. et al. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nature Neurosci. 4, 231–232 (2001).
Cai, H. et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nature Neurosci. 4, 233–234 (2001).
Roberds, S. L. et al. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum. Mol. Genet. 10, 1317–1324 (2001).
Turner, R. T. et al. Subsite specificity of memapsin 2 (β-secretase): implications for inhibitor design. Biochemistry 40, 10001–10006 (2001).
Kitazume, S. et al. Alzheimer's β-secretase, β-site amyloid precursor protein cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc. Natl Acad. Sci. USA 98, 13554–13559 (2001).
Kitazume, S. et al. Characterization of α2,6-sialyltransferase cleavage by Alzheimer's β-secretase (BACE1). J. Biol. Chem. 278, 14865–14871 (2003).
Lichtenthaler, S. et al. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 278, 48713–48719 (2003).
Luo, Y. et al. BACE1 (β-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time. Neurobiol. Dis. 14, 81–88 (2003).
Harrison, S. M. et al. BACE1 (β-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol. Cell. Neurosci. 24, 646–655 (2003).
Hsiao, K. et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996).
Ohno, M. et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41, 27–33 (2004).
John, V., Beck, J. P., Bienkowski, M. J., Sinha, S. & Heinrikson, R. L. Human β-secretase (BACE) and BACE inhibitors. J. Med. Chem. 46, 4625–4630 (2003).
Leung, D., Abbenante, G. & Fairlie, D. P. Protease inhibitors: current status and future prospects. J. Med. Chem. 43, 305–341 (2000).
Hong, L. et al. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 290, 150–153 (2000). Crystal structure of β-secretase, which was crucial for rational inhibitor design.
McGeer, P. L. & McGeer, E. G. Inflammation, autotoxicity and Alzheimer's disease. Neurobiol. Aging 22, 799–809 (2001).
McGeer, P. L., Schulzer, M. & McGeer, E. G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiological studies. Neurology 47, 425–432 (1996).
Rogers, J. et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology 43, 1609–1611 (1993).
Sainali, S. M., Ingram, D. M. & Talwaker, S. in 6th International Stockholm-Springfield Symposium on Advances in Alzheimer Therapy Abstr. (Stockholm, 2000).
Aisen, P. S. et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826 (2003).
Lim, G. P. et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J. Neurosci. 20, 5709–5714 (2000).
Yan, Q. et al. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J. Neurosci. 23, 7504–7509 (2003).
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S. & Drachman, D. A. Statins and the risk of dementia. Lancet 356, 1627–1631 (2000).
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G. G. & Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443 (2000).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 (1993).
Sing, C. F. & Davignon, J. Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am. J. Hum. Genet. 37, 268–285 (1985).
Wolozin, B. Cholesterol and the biology of Alzheimer's disease. Neuron 41, 7–10 (2004).
Puglielli, L. et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nature Cell Biol. 3, 905–912 (2001).
Adamson, P. & Greenwood, J. How do statins control neuroinflammation? Inflam. Res. 52, 399–403 (2003).
Jonhagen, M. E. et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer's disease. Dementia Geriat. Cogn. Disord. 9, 246–257 (1998).
Tuszynski, M. H. et al. in 56th Annual Meeting of the American Academy of Neurology abstr. S17.002 (San Francisco, USA, 2004).
Moller, H. J. Reappraising neurotransmitter based strategies. Eur. Neuropsychopharmacol. 9 (Suppl.), 53–59 (1999).
Gill, S. S. et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson's disease. Nature Med. 9, 589–595 (2003).
Arriagada, P. V., Growdon, J. H., Hedley-White, E. T. & Hyman, B. T. Neurofibrillary tangles, but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639 (1992).
Lee, V. M. Y. & Trojanowski, J. Q. Neurodegenerative tauopathies: human disease and transgenic mouse models. Neuron 24, 507–510 (1999).
Lewis, J. et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nature Genet. 25, 402–405 (2000).
Noble, W. et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565 (2003).
Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 (1991).
Author information
Authors and Affiliations
Ethics declarations
Competing interests
M. Citron is an employee of Amgen Incorporated.
Glossary
- NUCLEUS BASALIS
-
A telencephalic nucleus that is the main provider of cortical acetylcholine.
- NEUROPIL
-
A felt-like network that is interspersed between the cells of the grey matter in the CNS. It consists of neuronal and glial processes and synaptic terminals.
- ADJUVANT
-
An agent mixed with an antigen that enhances the immune response to that antigen upon immunization.
- PASSIVE IMMUNIZATION
-
The induction of immunity by the transfer of immunoglobulins.
- MICROGLIA
-
Phagocytic immune cells in the brain that engulf and remove cells that have undergone apoptosis.
- F(AB')2 FRAGMENTS
-
Dimers of the antigen-binding portion of an antibody.
- PDAPP MICE
-
A mouse line that is genetically altered to develop amyloid plaques.
- MENINGOENCEPHALITIS
-
An inflammatory process involving the brain and meninges, most often produced by pathogenic organisms that invade the central nervous system, and occasionally by toxins, autoimmune disorders and other conditions.
- LEPTOMENINGES
-
The collective term for the pia mater and arachnoid layers of the meninges.
Rights and permissions
About this article
Cite this article
Citron, M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci 5, 677–685 (2004). https://doi.org/10.1038/nrn1495
Issue Date:
DOI: https://doi.org/10.1038/nrn1495
This article is cited by
-
Quantitative assessment of AD markers using naked eyes: point-of-care testing with paper-based lateral flow immunoassay
Journal of Nanobiotechnology (2021)
-
Curcumin inhibits BACE1 expression through the interaction between ERβ and NFκB signaling pathway in SH-SY5Y cells
Molecular and Cellular Biochemistry (2020)
-
Meta-analysis on big data of bioactive compounds from mangrove ecosystem to treat neurodegenerative disease
Scientometrics (2020)
-
Delivery of BACE1 siRNA mediated by TARBP-BTP fusion protein reduces β-amyloid deposits in a transgenic mouse model of Alzheimer’s disease
Journal of Biosciences (2019)
-
Role of BACE1 in Alzheimer’s synaptic function
Translational Neurodegeneration (2017)
