
Key Points
-
The nuclear factor-κB (NF-κB) family of transcription factors plays a central part in orchestrating the innate and adaptive immune response against invading microbial pathogens. Successful pathogens have acquired multiple mechanisms to control the signalling pathways that regulate the NF-κB cascade, and they can collectively target every aspect of the NF-κB signalling pathway. This Review focuses on the different strategies used by viruses, bacteria and parasites to manipulate NF-κB functions.
-
The essential role of NF-κB family members in protection against viruses, bacteria and parasites has been established by the observation that knocking down (ablating) the expression of any of the NF-κB components or blocking their signalling function increases host susceptibility to diverse microbial pathogens.
-
Viruses are obligate intracellular parasites and frequently either activate or inhibit the NF-κB signalling pathway during different phases of their life cycle. So, a delicate balance must be maintained between activation and inhibition of the NF-κB pathway. Some viruses uniquely activate NF-κB to promote viral replication and to control virus-induced cellular apoptosis.
-
Viruses usually activate or suppress the NF-κB signalling pathway by modulating the function of multiple signalling receptors, adaptor molecules, cellular kinases, and cellular NF-κB inhibitors or regulators, and also by direct interaction with the NF-κB subunits themselves. Virus-encoded proteins can also sometimes modulate the transcriptional activity of activated NF-κB after translocation to the nucleus.
-
Bacteria and parasites can activate or inhibit the NF-κB signalling pathway, depending on the host cellular status that most benefits the pathogen. Bacteria-derived molecules can modulate NF-κB by targeting the same pathway components and stages as are targeted by viruses. Sometimes, bacteria and parasites can hijack crucial NF-κB signalling molecules and re-appropriate them for exploitation during the pathogen's own replication cycle.
-
Although virus- and bacteria-derived molecules are remarkably diverse in structure, they often target operationally common NF-κB signalling molecules for modulation. Interestingly, a single pathogen often encodes multiple proteins to modulate the NF-κB signalling pathway at different steps, which affirms the key importance of the NF-κB pathway in combating diverse invading microbial pathogens.
Abstract
The nuclear factor-κB (NF-κB) family of transcription factors plays a central part in the host response to infection by microbial pathogens, by orchestrating the innate and acquired host immune responses. The NF-κB proteins are activated by diverse signalling pathways that originate from many different cellular receptors and sensors. Many successful pathogens have acquired sophisticated mechanisms to regulate the NF-κB signalling pathways by deploying subversive proteins or hijacking the host signalling molecules. Here, we describe the mechanisms by which viruses and bacteria micromanage the host NF-κB signalling circuitry to favour the continued survival of the pathogen.
Similar content being viewed by others

Main
The nuclear factor-κB (NF-κB) family of transcription factors regulates the expression of hundreds of genes that are associated with diverse cellular processes, such as proliferation, differentiation and death, as well as innate and adaptive immune responses. The mammalian NF-κB proteins are members of the Rel domain-containing protein family: RELA (also known as p65), RELB, c-REL, the NF-κB p105 subunit (also known as NF-κB1; which is cleaved into the p50 subunit) and the NF-κB p100 subunit (also known as NF-κB2; which is cleaved into the p52 subunit); these proteins can homodimerize or heterodimerize through their conserved Rel homology domain to mediate gene transcription1,2. NF-κB proteins are activated by a variety of diverse extracellular or intracellular stimuli, including microbial pathogens and pathogen-associated molecular patterns (PAMPs)3. The NF-κB signalling pathway is an attractive target for exploitation by microbial pathogens in order to modulate host cell events, as activation of NF-κB is such a rapid response. Cytoplasmic NF-κB complexes are transferred to the nucleus within minutes after exposure to a pathogen or PAMPs, even in the absence of de novo protein synthesis, and induce the expression of a broad spectrum of antimicrobial pro-inflammatory cellular response genes4,5. The central role of these transcription factors in pathogen defence is highlighted by the fact that the NF-κB signalling cascade is probably the most frequently targeted intracellular pathway for subversion by anti-immune modulators that are encoded by a wide spectrum of microbial pathogens4. In this Review, we describe some of the recent advances in our understanding of the various mechanisms used by pathogens to modulate NF-κB signalling.
Signalling targets upstream of NF-κB
NF-κB proteins are tightly regulated in both the cytoplasm and the nucleus6. Under normal physiological conditions, NF-κB complexes remain inactive in the cytoplasm through a direct interaction with proteins of the inhibitor of NF-κB (IκB) family, including IκBα, IκBβ and IκBɛ (also known as NF-κBIα, NF-κBIβ and NF-κBIɛ, respectively); IκB proteins mask the nuclear localization domains in the NF-κB complex, thus retaining the transcription complex in the cytoplasm. In response to diverse stimuli, various cellular immune receptors (such as Toll-like receptors (TLRs)) and cytokine receptors (such as the interleukin-1 receptors (IL-1Rs), TNF receptors (TNFRs) and other TNFR-like receptors) can rapidly activate the NF-κB complex following the appropriate pro-inflammatory stimulation. This activation is mediated by a signalling cascade that uses multiple adaptors (including TNFR-associated factors (TRAFs), myeloid differentiation primary response protein 88 (MYD88) and TIR domain-containing adaptor protein (TIRAP)), as well as intermediate transducing molecules and kinases (including IL-1R-associated kinases (IRAKs), receptor-interacting proteins (RIPs; also known as RIPKs) and NF-κB-inducing kinase (NIK; also known as MAP3K14)), to eventually lead to degradation of the inhibitory protein IκBα, thus liberating the NF-κB complexes for transport to the nucleus, where they undergo further layers of regulation7 (Fig. 1).

Protein levels and activity of signalling molecules can be regulated through post-translational modifications such as phosphorylation, ubiquitylation and acetylation. The activation of nuclear factor-κB (NF-κB) ultimately results in the transcription of genes that encode pro-inflammatory factors and factors that influence cell proliferation. IκBα, NF-κB inhibitor-α (also known as NF-κBIα); IKK, IκB kinase; IL-1R, interleukin-1 receptor; NEMO, NF-κB essential modulator (also known as IKKγ); NIK, NF-κB-inducing kinase (also known as MAP3K14); TLR, Toll-like receptor; TNFR, TNF receptor.
The receptor-mediated signalling events converge on the same core components of the NF-κB activation apparatus: the IκB kinase (IKK) complex, which is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, NEMO (NF-κB essential modulator; also known as IKKγ)8,9. Activation of the IKK complex induces phosphorylation of IκBα, followed by ubiquitylation by a specific cullin-RING ubiquitin ligase (CRL) family SCFβTRCP complex, which contains SKP1 (S phase kinase-associated protein 1), cullin 1 and the F-box protein βTRCP as substrate adaptor; this SCFβTRCP complex specifically recognizes and degrades IκBα that is phosphorylated on Ser32 and Ser36. As well as processing IκBα, the ubiquitylation–proteasome pathway is important for the processing of NF-κB subunits p105 and p100 to p50 and p52, respectively10. In addition to the IKK complex, members of the IKK family of kinases — including IKKi (inducible IKK; also known as IKKɛ) and TBK1 (TANK-binding kinase 1) — can activate the NF-κB pathway in response to microbial infection11. TBK1 interacts with TANK, a TRAF-binding protein that activates NF-κB by modulating the function of TRAF2 and that also interacts with IKKi. TBK1 enhances the enzymatic activity of IKKβ by direct phosphorylation, and thus contributes to NF-κB activation12. Because of the essential role of IKKs and IκBα in the activation and regulation of NF-κB signalling, these proteins (or their post-translational modifications) are often targeted directly by microbial pathogens to control the host immune responses.
The NF-κB pathway is classified as either classical (canonical) or alternative (non-canonical) on the basis of the IKK subunits that get activated by upstream kinases13,14 (Fig. 1). In the classical pathway (for example, triggered by TNFR1 signalling), IKKβ and NEMO become activated by adaptors (such as TRAFs) and then phosphorylate p105 and IκBα to release the prototypical heterodimer p50–RELA. In the alternative pathway (for example, triggered by lymphotoxin-β receptor), IKKα is activated by NIK and then phosphorylates p100, which is subsequently cleaved to form p52. p52 then forms a heterodimer with RELB and translocates to the nucleus. This alternative pathway is triggered by a subset of tumour necrosis factor (TNF) family members, including CD40, lymphotoxin-β, B cell-activating factor (BAFF), receptor activator of NF-κB ligand (RANKL) and TNF-related weak inducer of apoptosis (TWEAK). By contrast, inflammatory cytokines, genotoxic stress, antigens and TLR stimulation tend to activate the classical pathway. Both the classical and alternative pathways are modulated by microbial pathogens, as the two pathways induce coordinated immune responses following diverse infections.
Outcome of NF-κB activation
Activation of NF-κB is considered to be the central initiating cellular event of host responses to invasion by microbial pathogens. The presence of a functional NF-κB signalling cascade in the horseshoe crab, a species that is known as a 'living fossil', suggests that the proteins involved are the evolutionarily conserved immune defence molecules15, and highlights the central role of NF-κB in upregulating the expression of genes encoding chemokines, cytokines, adhesion molecules (such as intercellular adhesion molecule 1 (ICAM1)), enzymes that produce secondary inflammatory mediators, and inhibitors of apoptosis. These molecules are key components of the innate immune response to invading microorganisms and are required for the migration of inflammatory and phagocytic cells to the site of infection, where NF-κB has been activated. The activated phagocytic cells kill, ingest and degrade microbial pathogens and eventually present the antigens to T cells after they re-migrate to secondary lymphoid organs. The secreted cytokines, including TNF and IL-1β, also start a feedback loop for a second phase of NF-κB activation that continues the induction of robust immune responses. The cellular pattern recognition receptors (PRRs) such as TLRs, RIG-I-like receptors (RLRs) and NOD-like receptors (NLRs), which all sense microbial pathogens and PAMPs, use distinct signalling pathways that eventually converge to activate NF-κB16, leading to the production of inflammatory mediators.
The initiation of innate immune events is important in host resistance to many different types of pathogens prior to the activation of adaptive immune responses, which are also mediated by NF-κB. In addition, NF-κB plays a part in the suppression of apoptosis induced by bacterial components such as lipopolysaccharide. The importance of NF-κB in protection against pathogens is also supported by the fact that NF-κB complexes are abundantly expressed in myeloid cells, which are mostly dedicated to innate immunity. The importance of members of the NF-κB family in both the immune response against pathogens and the development and function of other immune cells has been demonstrated by sequentially knocking down the expression levels of these proteins17. Mice lacking different NF-κB family members become susceptible to various viral, bacterial and parasitic infections (Table 1). In addition, IKK and IKK-related kinases also play an important part in regulating antimicrobial responses11,18. As a result, pathogens have developed various mechanisms to alter the activation of NF-κB. Here, we focus on the mechanisms by which viruses and bacteria modulate NF-κB activation.
Modulation of NF-κB by viruses
NF-κB-dependent genes are important for regulating cellular processes such as apoptosis, inflammation and immune responses, and so many viruses have acquired diverse strategies to regulate NF-κB signalling19. Some viruses regulate NF-κB in a biphasic manner to optimize viral replication in the infected cells during different phases of the viral life cycle. For example, some viruses (such as the gammaherpesviruses Kaposi's sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV)) activate NF-κB during latency, whereas they tend to suppress NF-κB signalling during the lytic cycle20. By contrast, viruses such as Sindbis virus, dengue virus and reoviruses activate NF-κB to facilitate the induction of apoptosis, which increases viral spread via phagocytic myeloid cells. However, more often, viruses inhibit NF-κB primarily to dampen host inflammatory and immune responses, often by encoding multiple proteins that target the pathways at multiple levels21,22,23. Larger DNA viruses, such as poxviruses, frequently deploy several proteins to regulate NF-κB in cultured cells in a seemingly redundant manner, whereas smaller RNA viruses may encode only a single protein that has multiple roles in regulating NF-κB. Tables 2,3 list some of the NF-κB modulators that are derived from viruses.
Activation of NF-κB by viruses
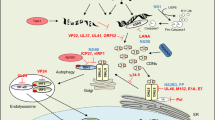
Some viruses have evolved mechanisms to pro-actively stimulate NF-κB activation19,21,24. Viruses such as HIV-1, human T-lymphotrophic virus 1 (HTLV-1), hepatitis B virus (HBV), hepatitis C virus (HCV), rotaviruses, influenza viruses and respiratory syncytial viruses (RSVs) activate NF-κB to promote viral replication and to pre-empt virus-induced apoptosis (Fig. 2). These viral genomes all possess strategic gene promoters with NF-κB-binding sites and, thus, activated NF-κB is crucial for viral gene expression, replication and spread. Infections with RSVs, the causative agents of acute respiratory diseases such as bronchiolitis and pneumonia, are associated with excessive inflammation caused by the release of NF-κB-regulated pro-inflammatory cytokines and chemokines by airway epithelial cells25. This occurs because the infection induces a persistent activation of both classical and alternative NF-κB pathways25. The classical NF-κB pathway is activated by the host superoxide-generating enzyme NOX2-containing NADPH oxidase, which phosphorylates IκBα and RELA in airway epithelial cells via RIG-I (also known as DDX58), TRAF6 and IKKβ26. However, the alternative NF-κB pathway is activated by the kinases NIK and IKKα, leading to the nuclear translocation of p52–RELB27. This suggests that the redox modification mediated by RSVs might have multiple upstream targets that then lead to the activation of both NF-κB pathways. Two of the RSV proteins, fusion glycoprotein F and M2-1, have been shown to activate NF-κB-mediated cytokine induction in monocytic cells28,29.

A diagrammatic representation of the nuclear factor-κB (NF-κB) pathways that are induced by various receptors, microbial pathogens and pathogen-derived proteins. See main text for details. Virus-encoded proteins are in green boxes, and bacterium- and parasite-encoded proteins are in orange boxes. ASFV, African swine fever virus; BFV, bovine foamy virus; B. pertussis, Bordetella pertussis; CRL, cullin-RING ubiquitin ligase; EBV, Epstein–Barr virus; FHA, filamentous haemagglutinin; gp350, glycoprotein 350; HBV, hepatitis B virus; HCV, hepatitis C virus; HSV, herpes simplex virus; HSVA, herpesvirus ateles; HSVS, herpesvirus saimiri; HTLV-1, human T cell leukaemia virus 1; IκBα, NF-κB inhibitor-α (also known as NF-κBIα); IKK, IκB kinase; IRAK, IL-1R-associated kinase; KSHV, Kaposi's sarcoma-associated herpesvirus; LMP1, latent membrane protein 1; L. pneumophila, Legionalla pneumophila; MYD 88, myeloid differentiation primary response protein 88; NEMO, NF-κB essential modulator (also known as IKKγ); RIP1, receptor-interacting protein 1 (also known as RIPK1); R. rickettsii, Rickettsia rickettsii; RSV, respiratory syncytial virus; SCFβTRCP, SKP1, cullin 1 and F-box protein βTRCP; S. flexneri, Shigella flexneri; Stp, Saimiri transformation-associated protein; TAK1, TGFβ-activated kinase 1; T. gondii, Toxoplama gondii; TLR, Toll-like receptor; TNFR, TNF receptor; TRAF, TNFR-associated factor; Ub, ubiquitin.
The HIV-1 proteins Tat, Vpr and Nef activate the NF-κB pathway by exploiting multiple mechanisms. Tat, a transcriptional activator, induces transcription of cytokines such as IL-10, IL-6 and TNF in monocytes or macrophages; these cytokines promote HIV-1 replication at early stages of infection30. Tat also increases the DNA-binding activity of NF-κB complexes by promoting acetylation of p50 via the CREBBP–p300 complex31. However, recent studies have demonstrated that Tat also can inhibit NF-κB activation, as discussed below. Vpr, a viral late protein, and Nef, a viral early protein, enhance virion infectivity and increase viral replication. These viral proteins can activate the NF-κB pathway when added exogenously and stimulate the transcription of HIV-1 genes in promonocytic cells and primary macrophages32,33.
Many oncogenic viruses activate NF-κB to facilitate transformation of infected cells. In most cases, virus-encoded oncoproteins are directly involved in this activation process. Among the oncoproteins identified so far, the mechanisms of persistent NF-κB activation by Tax1 from HTLV-1 and Tax2 from HTLV-2 during T cell leukaemia are well established34,35. Tax1 primarily activates the IKK complex by directly interacting with the non-catalytic subunit, NEMO36. In addition, Tax1 modulates the activity of kinases that activate the IKK complex, such as MEK kinase 1 (MEKK1; also known as MAP3K1), NIK and TGFβ-activated kinase 1 (TAK1; also known as MAP3K7) (Ref. 37). Recently, it was demonstrated that Tax1 sequesters the activated IKK complex in lipid rafts through its interaction with NEMO, providing an optimal microenvironment for kinase activation and thus allowing constitutive activation of the NF-κB pathway38. Unlike Tax2, Tax1 also modulates the alternative NF-κB pathway by upregulating IKKα in T cells, thereby inducing the processing of p100 (Ref. 39). Another member of the Retroviridae, bovine foamy virus (BFV), activates the classical and alternative NF-κB pathways using the viral transactivator BTas, which interacts with IKKα and IKKβ and persistently activates NF-κB40.
EBV, a human gammaherpesvirus that causes multiple types of cancer, is a strong inducer of NF-κB activation. During the early phase of infection, the binding of EBV glycoprotein gp350 (or its alternative isoform, gp250) to the cellular receptors CD21 (also known as CR2) and TLR2 causes persistent activation of the classical NF-κB pathway41. At later stages of EBV infection, B cells are immortalized, in part owing to the activation of NF-κB by viral latent membrane protein 1 (LMP1), which mimics a constitutively activated TNFR42. LMP1 activates both the alternative and the classical NF-κB pathways using its carboxy-terminal activation region 1 (CTAR1) and CTAR2, respectively43,44. CTAR2 interacts with TRAF6, TRADD and RIP1, which activate IKKβ and NEMO45, thus activating the classical NF-κB pathway, whereas CTAR1 interacts with TRAF2 and TRAF3, which activate NIK and IKKα, thus activating the alternative pathway46; in fact, the identification of these signalling molecules using LMP1 led to the identification of the alternative NF-κB pathway. However, recent studies suggest that additional mechanisms are involved in LMP1-mediated activation of NF-κB47.
Similarly to EBV, KSHV, another human oncogenic gammaherpesvirus, establishes latent infection by activating NF-κB. The K13 protein, known as vFLIP, is one of the proteins that regulates the latency of KSHV. vFLIP interacts with NEMO in the IKK complex to activate classical NF-κB signalling48. In addition, vFLIP blocks KSHV lytic replication by antagonizing the KSHV lytic genes, including RTA (replication and transcription activator) and vGPCR, by binding to their promoters49. Moreover, activation of NF-κB by vFLIP suppresses the host AP1 pathway, which is essential for KSHV lytic replication50, and upregulates the expression of miR-146a, which suppresses the expression of CXCR4, a CXC-chemokine receptor for stromal-derived factor 1 (SDF1; also known as CXCL12); the suppression of CXCR4 may enhance the spread of KSHV-infected endothelial cells51. Thus, vFLIP has multiple roles in the regulation of cellular gene expression in order to maintain the fine balance between latency and lytic replication.
Some gammaherpesviruses that cause lymphoma in New World primates harbour several oncoproteins that activate NF-κB and transform human T cells. The oncoproteins Saimiri transformation-associated protein C (StpC) and StpA11 of herpesvirus saimiri induce TRAF2 and TRAF6 and activate both NF-κB pathways52,53,54. Tio, an oncoprotein from herpesvirus ateles (a virus that causes T cell malignancies in primates), also activates both NF-κB pathways using TRAF6 to activate the IKK complex54.
Some viruses modulate the kinases associated with the NF-κB pathway to promote transcription of their own genes. Murine gammaherpesvirus 68 (MuHV-68), which establishes long-term latent infection in the mouse spleen, initiates lytic replication using its RTA protein (encoded by ORF50) in latently infected cells. MuHV-68 activates IKKβ in a MAVS (mitochondrial antiviral-signalling protein)-dependent manner through phosphorylation of RTA, to promote the lytic replication cycle55, although a previous study reported that the NF-κB pathway is dispensable for MuHV-68 lytic replication56. HBV, which causes hepatocellular carcinoma (the most common form of liver cancer in adults), can persistently activate NF-κB using the transcriptional transactivator protein HBx (also known as protein X)57. Several mechanisms have been proposed for the induction of NF-κB target genes by HBx, including direct targeting of the NF-κB components and, as an indirect route, activation of cellular kinases that induce transcription of cellular genes containing κB elements (sequences to which NF-κB binds) in their promoters58. A complex consisting of HBx and RELA can activate the gene encoding metastasis-associated protein 1 (MTA1), which is a master chromatin modifier, and can regulate cancer progression, indicating the importance of NF-κB upregulation59. HCV, which causes acute and chronic hepatitis, also persistently activates NF-κB, leading to liver cirrhosis and hepatocellular carcinoma. The core and NS3 proteins of HCV activate NF-κB by activating TNFR1 and TLRs60. Apart from using oncoproteins, viruses use homologues of cellular anti-apoptotic proteins as a survival strategy; for example, the African swine fever virus (ASFV) inhibitor of apoptosis (IAP), A224L, can activate NF-κB using TRAF2 and IKKβ, and can thus prevent apoptosis61.
Other viruses, such as herpes simplex virus (HSV) spp. and HIV-1, activate NF-κB in a biphasic manner for viral infection and replication. The early phase of NF-κB activation happens independently of viral replication, whereas the second phase requires viral gene expression. In the case of HSV, the early phase of NF-κB activation is mediated by envelope glycoprotein D and involves the host protein herpesvirus entry mediator A (HVEA)62 and the viral tegument protein UL37, which binds to TRAF6 during entry63. After entry, the HSV immediate–early proteins ICP4 and ICP27 start the second phase of NF-κB activation in a TLR-independent manner64.
Suppression of receptors and NF-κB adaptors
Viruses suppress NF-κB activation to dampen the host immune responses and, in some cases, to maintain latency. Multiple virus-encoded proteins have been identified that inhibit NF-κB activation by targeting the inducer ligands, receptors or sensors that activate innate responses, the downstream adaptor molecules in these pathways, or the kinases that activate the NF-κB pathways (Fig. 3). As mentioned above, some viruses (for example, HSV spp.), maintain a delicate balance between activation and suppression of NF-κB in order to maintain long-term persistence. Activation of NF-κB is required to start the infection and viral replication; however, NF-κB signalling also promotes the expression of inflammatory cytokines, and several viral proteins — the immediate–early proteins of HSV, for example — block NF-κB activation at a later stage to ensure a smooth progression of infection. The HSV immediate–early protein ICP0, an E3 ubiquitin ligase, reduces TLR2-mediated inflammatory responses against the virus by inducing degradation of the adaptor protein MYD88 (Ref. 65), whereas ICP27, another immediate–early protein, represses NF-κB function by stabilizing IκBα through blockade of its phosphorylation and ubiquitylation66. This suggests that bifunctional viral proteins such as ICP27, which activate as well as suppress NF-κB, have complex regulatory duties that function to maintain a delicate balance between advantageous and deleterious host responses.

A diagrammatic representation of the nuclear factor-κB (NF-κB) pathways, showing the signalling molecules that are targeted by microbial pathogen-derived proteins for inhibition of the NF-κB pathways. See main text for details. Virus-encoded proteins are in green boxes, and bacterium-encoded proteins are in orange boxes. Ac, acetyl group; A. salmonicida, Aeromonas salmonicida; ASFV, African swine fever virus; B. bronchiseptica, Bordetella bronchiseptica; ChlaDub1, Chlamydia deubiquitylase and deneddylase; CPXV, cowpox virus; CRL, cullin-RING ubiquitin ligase; C. trachomatis, Chlamydia trachomatis; EBV, Epstein–Barr virus; EHEC, enterohaemorrhagic Escherichia coli; EPEC, enteropathogenic E. coli; HAdV-12, human adenovirus 12; HaV, hantaan virus; HCMV, human cytomegalovirus; HCV, hepatitis C virus; HSV, herpes simplex virus; IκBα, NF-κB inhibitor-α (also known as NF-κBIα); IKK, IκB kinase; IRAK, IL-1R-associated kinase; M, membrane protein; MOCV, molluscum contagiosum virus; MYD88, myeloid differentiation primary response protein 88; MYXV, myxoma virus; N, nucleocapsid protein; NEMO, NF-κB essential modulator (also known as IKKγ); PPV, parapoxvirus; RIP1, receptor-interacting protein 1 (also known as RIPK1); RPS3, 40S ribosomal protein S3; RV, rotavirus; SARS-CoV, SARS coronavirus; S. boydii, Shigella boydii; SCFβTRCP, SKP1, cullin 1 and F-box protein βTRCP; S. flexneri, Shigella flexneri; S. Typhimurium, Salmonella enterica subsp. enterica serovar Typhimurium; TAK1, TGFβ-activated kinase 1; TLR, Toll-like receptor; TNFR, TNF receptor; TRAF, TNFR-associated factor; Ub, ubiquitin; VACV, vaccinia virus; VARV, variola virus; V. parahaemolyticus, Vibrio parahaemolyticus.
HCV establishes persistent intra-hepatic infection using multiple proteins to either activate NF-κB and stimulate viral replication, or inhibit NF-κB and suppress the expression of its target host defence genes. The NS3–NS4A protease of HCV causes proteolysis of TRIF (TIR domain-containing adaptor inducing IFNβ; also known as TICAM1), an adaptor protein used by TLR3 and MAVS, and this proteolysis inhibits NF-κB and interferon regulatory factor 3 (IRF3)67,68. Another HCV protein, phosphoprotein NS5A, interacts with MYD88 and TRAF2 in macrophage cell lines and inhibits the TLR2-, TLR4-, TLR7- and TLR9-mediated activation of NF-κB69. However, NS5A from bovine viral diarrhoea virus, a member of the Flaviviridae that is closely related to HCV, modulates host immune responses by interaction with NIK- and IKKβ-binding protein (NIBP; also known as TRAPPC9) in LB9.K cells70, suggesting that related viral immunomodulatory molecules might have multiple cellular targets to regulate NF-κB in different ways, depending on the host. Among the other HCV proteins, NS5B and the core proteins interact with IKKs and inhibit activation of the IKK complex71,72.
Viruses frequently target IKKs to mediate NF-κB inhibition, as diverse signalling pathways converge on these kinases. SARS coronavirus (SARS-CoV), which causes life-threatening atypical pneumonia, modulates signalling through IKK and subsequently interrupts NF-κB activation via viral membrane (M) protein73. M protein physically interacts with IKKβ, thereby suppressing TNF-induced activation of NF-κB and the subsequent expression of cyclooxygenase 2, an enzyme that is known to have an antiviral function73. In a similar manner, the EBV protein EBNA1 inhibits phosphorylation of IKKα–IKKβ to suppress the classical NF-κB pathway in carcinoma cells74.
Members of the Poxviridae, a large family of DNA viruses, modulate NF-κB function through multiple proteins that target diverse NF-κB signalling molecules, including the IKK complex75. Vaccinia virus (VACV), the prototypical orthopoxvirus, encodes a remarkable NF-κB tool kit that includes multiple inhibitory proteins — such as A46R, A52R, B14, K1L M2L and N1L — all of which block activation of the IKK complex and inhibit degradation of IκBα, albeit using diverse mechanisms76,77,78,79. Among these, B14 inhibits NF-κB by directly targeting IKKβ of the IKK complex80. Interestingly, the B14 counterpart in the attenuated virus modified virus Ankara (MVA), encoded by ORF183 and lacking six amino acids that are present in α-helix 6 of B14, cannot inhibit NF-κB activation81; as a result, MVA infection activates NF-κB, whereas VACV infection inhibits NF-κB very quickly after activating it. The VACV proteins E3L, K1L and N1L use multiple targets, as they all block both NF-κB and antiviral pathways82,83. Molluscum contagiosum virus (MOCV) protein MC159 is a vFLIP that inhibits FAS-mediated apoptosis and TNF-induced late activation of NF-κB. However, transgenic expression of this protein in mice instead enhanced NF-κB-mediated immune responses84,85. Another MOCV protein, MC160, inhibits NF-κB activation using multiple mechanisms86. ORF024 of the parapoxviruses encodes a unique protein that modulates the NF-κB induction pathway. In the absence of this gene, the virus replicates normally in primary OFTu cells, but induces higher expression of NF-κB-regulated chemokines and other pro-inflammatory host genes than the wild-type virus87. The ORF024 protein expressed by itself decreased phosphorylation of IKK and the downstream activation of the NF-κB pathway, suggesting that this viral protein targets upstream kinases that phosphorylate IKK.
Targeting NF-κB and its cellular inhibitors
Viruses can also regulate the function of NF-κB complexes through proteins that either directly interact with the nuclear factors themselves or control the cellular regulators of these factors. This indirect regulation includes inhibition of IκBα degradation, of dimer complex formation among the NF-κB members, of nuclear translocation or of binding to the targeted cellular gene promoters (Fig. 3). Some viruses encode homologues of cellular NF-κB signalling molecules, and these homologues act as dominant negatives of the cellular proteins. For example, ASFV encodes a homologue of IκBα, A238L, which interacts with cellular RELA and thus suppresses the activation of NF-κB complexes88. Furthermore, virus-encoded proteases can cleave IκBα and NF-κB subunits to render them non-functional. The human coxsackievirus B3 protease 3C cleaves IκBα to create an amino-terminal fragment that interacts with RELA and translocates with it to the nucleus, where the complex therefore remains inactive89. This blockade of NF-κB activation induces apoptosis of infected cells. Another protease, amino-terminal protease (Npro) from classical swine fever virus, also interacts with IκBα and modulates NF-κB function90.
Some viral proteins block NF-κB activation by preventing the degradation of IκBα. For example, HIV-1 modulates IκBα function in order to maintain long-term infection. The Vpu protein of HIV-1 blocks proteasome-dependent degradation of IκBα by binding to βTRCP in the E3 ubiquitin ligase complex that is involved in the regulated degradation of IκBα91,92. Thus, HIV-1 induces apoptosis of infected T cells by reducing the expression of NF-κB-dependent cellular anti-apoptotic factors such as BCL-XL and TRAF1. The rotaviral non-structural protein NSP1 induces proteasome-dependent degradation of βTRCP to stabilize IκBα and inhibit NF-κB93. Another HIV-1 protein, Tat, also inhibits degradation of IκBα and nuclear translocation of RELA94. This modulation of immune responses by Tat at late stages may provide a favourable environment for both HIV-1 and other opportunistic microorganisms. However, at early stages of infection, Tat instead activates NF-κB.
Some virus-encoded proteases can cleave NF-κB itself. For example, the poliovirus protease 3C cleaves RELA at the later stages of infection and thereby suppresses NF-κB activation95. Other virus-encoded proteins instead regulate the nuclear translocation of RELA. The cowpox virus ankyrin repeat domain-containing host range protein CP77 directly targets RELA and blocks its translocation to the nucleus to inhibit NF-κB96. Several other poxviral proteins suppress NF-κB function by preventing the degradation of the precursor molecule p105 (Refs 75, 97, 98). Hantaan virus, a member of the Bunyaviridae, uses its nucleocapsid (N) protein to block the nuclear translocation of RELA by interacting with importin-α proteins, which are nuclear transport proteins used by the NF-κB proteins to translocate to the nucleus99. The E1A protein from human adenovirus 12 associates with RELA and prevents PKAc, the catalytic subunit of protein kinase A enzymes, from phosphorylating RELA at Ser276; this then downregulates transcription of the genes encoding major histocompatibility complex (MHC) class I molecules100. Similarly, E1A binds to p50 and prevents its phosphorylation at Ser337 by PKAc101.
Other mechanisms of NF-κB modulation
Human cytomegalovirus (HCMV) modulates the NF-κB pathway at every stage of the viral life cycle, either by activation or inhibition. NF-κB is activated during the viral entry process, whereas HCMV successfully inhibits the NF-κB pathway after the initial infection. HCMV immediate–early protein IE86 inhibits binding of NF-κB to the promoters of interferon-β (IFNβ), cytokines and chemokines in response to TNF stimulation or virus infection102. However, IE86 does not block the nuclear translocation of NF-κB or directly interact with the subunits, suggesting that the protein might target host factors required for the transcriptional activity of NF-κB. HCMV can also inhibit the activation of the NF-κB pathway that is induced by TNF and IL-1β at later times post-infection, by inhibiting IKK activation using a late viral gene product103,104. An HCMV-encoded homologue of human IL-10 also inhibits NF-κB activation105. Like HCMV, reoviruses also modulate NF-κB in a manner that promotes the viral life cycle in the infected host. For example, reovirus 3 strain Dearing activates NF-κB to induce NF-κB-dependent apoptosis106. However, another strain of reovirus 3 (strain Abney) inhibits NF-κB at a later stage of infection to promote induction of apoptosis in HEK293 and primary cardiac myocytes. Infection of cells with this strain inhibits activation of NF-κB by external stimuli, an effect that requires the T3 S1 gene segment, which is also responsible for regulating apoptosis107.
For many virus-encoded proteins (Tables 2,3), the cellular targets are still unknown. For example, the West Nile virus protein NS1 inhibits TLR3-mediated activation of the NF-κB pathway by blocking nuclear translocation of RELA and IRF3 in order to suppress the production of IL-6 and IFNβ108. KSHV-encoded miRNAs, which are expressed during viral latency and in Kaposi's sarcoma tumours, modulate cellular gene functions and are thought to play a part in the pathogenesis of KSHV-induced malignancies. Recently, it has been demonstrated that the deletion of 14 miRNA clusters from KSHV reduced virus-induced NF-κB activation and enhanced lytic induction, as KSHV miRNAs activate NF-κB and induce enhanced expression of RTA and major capsid protein109.
Modulation of NF-κB by bacteria
Bacterial proteins that directly interact with the NF-κB signalling pathway. Bacteria generally encode a larger repertoire of proteins than viruses, and many of these proteins are also predicted to have host modulatory functions110. Like viruses, bacteria modulate the NF-κB signalling pathway by either activation or inhibition, according to the requirement for the life cycle of the individual pathogen111. Individual bacteria often encode and deploy multiple effector proteins for regulation of the NF-κB signalling pathway. Recent studies suggest that functional redundancy exists among the effector proteins from a single strain of bacteria112. Table 4 lists some of the documented NF-κB modulators from bacteria and other microbial pathogens. Although the known bacteria-derived effector molecules that modulate the NF-κB pathway are not structurally related to the viral modulators, in many cases they target the same cellular signalling molecules (Figs 2,3). For example, viral and bacterial effector molecules target the same IKK complex, as well as IκBα, to inhibit the NF-κB pathway (Fig. 3). By contrast, structurally related bacterial effector molecules often have diverse cellular targets for the regulation of NF-κB function.
Unlike viruses, bacterial pathogens use secretion systems, which are multicomponent complexes that translocate virulence factors to the extracellular space or the cytosol of target eukaryotic cells. These secretion systems are grouped into seven classes (type I to type VII) according to their protein composition and their mechanism of function113. Together, these secretion systems transport hundreds of virulence factors and effector proteins, but only a few of these have been characterized in detail. The known effector proteins that modulate host innate immune responses associated with the NF-κB signalling pathway are primarily delivered by the type III secretion system (T3SS), as found in Salmonella spp. and Yersinia spp., or by the type IV secretion system (T4SS), as found in Bartonella spp. Type III effector proteins from Yersinia spp., known as Yops, counteract multiple signalling pathways that are activated in the infected host cells114. For example, YopJ (also known as YopP in Yersinia enterocolitica) inhibits NF-κB signalling to block the production of pro-inflammatory cytokines. However, multiple mechanisms of action were described for YopJ, as it has several protein targets in the NF-κB pathway. YopJ was identified as a cysteine protease that also has deubiquitylating and desumoylating activity; it removes polyubiquitin chains from IκBα and therefore inhibits proteasomal degradation of the protein115. YopJ can also remove ubiquitin chains from TRAF6 to inhibit TLR-mediated activation of NF-κB signalling116. In addition, it acts as an acetyltransferase that acetylates serine and threonine residues in the activation loops of IKKα and IKKβ, thereby blocking phosphorylation and activation of the IKK complex by upstream kinases117,118. Aeromonas salmonicida encodes AopP, a type III effector protein that is related to YopJ. Secretion of this effector prevents the nuclear translocation of RELA, but the direct host target of AopP is unknown. It does not inhibit the phosphorylation of IκBα, suggesting that AopP might prevent degradation of IκBα using a similar mechanism to that used by YopJ119. Thus, YopJ (variants of which are encoded by multiple Gram-negative bacterial species) has acquired several mechanisms of inhibiting the NF-κB signalling pathway.
Owing to the importance of the NF-κB signalling pathway in many crucial cellular processes, cells have developed multiple mechanisms to regulate the function of this pathway. One negative regulatory mechanism is deubiquitylation of signalling molecules that activate the IKK complex, by deubiquitylase enzymes such as CYLD and zinc finger protein A20 (also known as TNFAIP3) (Refs 120, 121). Several bacteria have co-opted this mechanism using virulence factors that deubiquitylate NF-κB signalling molecules. The type III effector protein AvrA of Salmonella enterica subsp. enterica serovar Typhimurium str. PhoPc, a non-pathogenic strain, is a deubiquitylase that is closely related to YopJ and that inhibits the NF-κB pathway by removing ubiquitin from IκBα and from β-catenin, a negative regulator of the pro-inflammatory NF-κB pathway in epithelial cells122,123. Another S. Typhimurium type III effector protein, SseL (encoded in Salmonella pathogenicity island 2), also possesses a deubiquitylase activity that inhibits degradation of IκBα124,125. S. Typhimurium lacking SseL causes increased NF-κB activation in macrophages as a result of ubiquitin-mediated degradation of IκBα.
Several bacterial proteins can inhibit the degradation of IκBα by targeting the cellular ubiquitin machinery. The Shigella flexneri T3SS effector OspG can modulate the host NF-κB function by blocking the degradation of phosphorylated IκBα, and thus blocking NF-κB activation, in response to TNF stimulation in and S. flexneri infection of epithelial cells126. OspG is a serine/threonine kinase that binds to various ubiquitylated ubiquitin-conjugating enzymes (E2s), including UBCH5 (also known as UBE2D1), to prevent ubiquitylation of phosphorylated IκBα126. NleH1 and NleH2 from Escherichia coli O157:H7 str. EDL9883, an enterohaemorrhagic E. coli (EHEC) strain, have a high level of sequence similarity with OspG but have different targets from OspG. NleH1 and NleH2 lack the ability to block IκBα degradation, but instead interact with the human 40S ribosomal protein S3 (RPS3), a subunit of NF-κB complexes that regulates NF-κB-dependent transcription127. This suggests that the bacterial effectors that regulate the NF-κB pathway have many different targets and substrates. The existence of multiple NF-κB inhibitors was observed in EHEC serogroup 0111, which encodes a homologue of OspG. Surprisingly, EHEC serogroup 0111 lacking OspG inhibited RELA transfer to the nucleus in response to TNF, suggesting that additional modulators from T3SS are present in this EHEC serogroup128.
Enteropathogenic E. coli (EPEC) can either activate or suppress NF-κB through T3SS-dependent translocation of effectors and T3SS-independent mechanisms, presumably by activation of TLRs129. The T3SS effector proteins NleE and NleB can inhibit NF-κB activation by inhibiting IκBα phosphorylation130; NleE and NleB block the phosphorylation and activation of IKKβ by targeting upstream molecules, such as TAK1 (Ref. 130). Furthermore, NleE can block nuclear translocation of c-REL, but not of p50 or the transcription factors STAT1 and STAT2, indicating that the block in nuclear translocation is specific for c-REL (Ref. 131). OspZ, the NleE homologue from S. flexneri 6 and Shigella boydii, also blocks nuclear translocation of RELA in response to TNF-mediated activation of NF-κB. Another S. flexneri effector protein, IpaH9.8, possesses E3 ligase activity and inhibits the NF-κB pathway through a unique mechanism: it interacts with the IKK regulatory subunit NEMO and with ABIN1 (also known as TNIP1), a ubiquitin-binding adaptor protein, to promote the ABIN1-dependent polyubiquitylation of NEMO. Subsequent degradation of NEMO inhibits the activation of NF-κB and thus downregulates the host inflammatory responses132.
E. coli K1 suppresses the production of pro-inflammatory cytokines from infected monocytes. However, in the absence of OmpA, the bacterium activates the NF-κB pathway via the extracellular signal-regulated kinase 1 (ERK1; also known as MAPK3)–ERK2 (also known as MAPK1) and the mitogen-activated protein kinase p38 (also known as MAPK14) pathways, resulting in the production of pro-inflammatory cytokines and chemokines133. Therefore, OmpA may target a kinase common to these pathways133. Opportunistic pathogens such as Pseudomonas aeruginosa synthesize a small molecule called N-(3-oxo-dodecanoyl) homoserine lactone (also known as C12), which inhibits the regulation of NF-κB functions in activated mammalian cells134. Modulation of IKK and inhibition of NF-κB signalling by C12 attenuates TLR4-dependent innate immune responses to promote persistent infection.
Like their viral counterpart proteins, bacterial effectors can inhibit the function of NF-κB transcription factors through direct interaction or proteolysis. Vibrio parahaemolyticus secretes the T3SS1-dependent effector protein VopS (encoded by the locus VP1686) into the cytosol of macrophages, and this then induces DNA fragmentation. VopS directly interacts with RELA to inhibit the DNA-binding activity of NF-κB, causing apoptosis of the infected macrophages135. The intracellular bacterial pathogen Chlamydia trachomatis, which infects human eyes and the urogenital tract, has acquired multiple mechanisms to modulate NF-κB function136. The C. trachomatis protein encoded by the locus CT441 (a Tsp-like protease) inhibits the nuclear translocation and function of NF-κB by cleaving RELA, and also inhibits NF-κB by regulating ubiquitin-mediated protein degradation137. Chlamydia deubiquitylase and deneddylase (ChlaDub1) binds IκBα and blocks its ubiquitylation and degradation, allowing C. trachomatis to evade the NF-κB-mediated host inflammatory response138. Chlamydia pneumoniae, which lacks ChlaDub1, uses an inclusion-specific protein (encoded by the locus CP0236) to sequester NF-κB activator (ACT1; also known as CIKS) and, thus, regulate NF-κB139.
Other bacterial proteins that affect NF-κB
Bacterial effector proteins can also activate the NF-κB pathway as a strategy of immune modulation (Fig. 2). Legionella pneumophila, which infects lung macrophages and causes Legionnaire's disease, activates NF-κB signalling in a T4SS-dependent manner. Using an NF-κB-specific luciferase reporter activation assay, LegK1 was identified as a potential activator of NF-κB signalling. LegK1, a eukaryotic-like serine/threonine kinase, potently and specifically activates host NF-κB signalling by directly phosphorylating IκBα and other members of the IκB family of inhibitors, as well as p100 (Ref. 140). Thus, LegK1 bypasses the requirement for host IKKs and upstream kinases, such as TRAF2, TRAF6, TAK1, NIK and MEKK3, in the activation of both classical and alternative NF-κB pathways. Rickettsia rickettsii, an obligate intracellular bacterial pathogen, activates IKKα and IKKβ to drive NF-κB activation in human endothelial cells141. Bordetella pertussis produces filamentous haemagglutinin (FHA), a cell-associated secreted adhesin that can induce early activation of the NF-κB pathway and cause the secretion of NF-κB-regulated inflammatory cytokines; however, longer exposure to this adhesin inhibits NF-κB activation, suggesting that there are complex temporal dynamics involved in the regulation of the innate response pathways142.
Like viruses, bacterial pathogens modulate host signalling to maintain a delicate balance between the cell death and survival pathways. For example, although infection of non-myeloid cells by S. flexneri induces inflammatory responses and activates anti-apoptotic pathways through the rapid activation of the NOD1-dependent RIP2–IKKβ–NF-κB signalling pathway, the bacterium can also induce apoptotic and necrotic cell death143. Thus, a balance between the induction of the apoptotic and anti-apoptotic pathways dictates the fate of the infected cell.
The intestinal microbiota of mammals and other metazoans maintain a homeostatic balance with the host immune system. Recent studies suggest that NF-κB signalling plays a crucial part in maintaining this host–bacteria symbiosis144. The host has adapted several mechanisms to distinguish between commensals and foreign pathogens, and exercises a delicate balance between tolerance and immunity. For example, in the intestine the expression pattern and localization of PRRs and the activation of NF-κB pathways have an important role in maintaining homeostasis144. Some commensal bacteria produce proteins that suppress immune activation; for example, Lactobacillus reuteri, a beneficial organism that is exploited for probiotics, can downregulate NF-κB-dependent host proteins that mediate cell proliferation and survival145. This species blocks nuclear translocation of RELA by preventing the degradation of IκBα in response to TNF stimulation145, although, as shown by a recent study using the commensal intestinal bacterium Lactobacillus plantarum, changes in host cell gene expression can depend on the bacterial growth phase146. On the other hand, the effects of probiotics in the prevention or treatment of diarrhoea caused by infection with enteric pathogens (for example, Saccharomyces boulardii) are thought to be mediated by immune modulation and the release of pro-inflammatory cytokines that are regulated by NF-κB147. This suggests that the commensals could compete with invading pathogens and enhance host defences.
Bacteria can also exploit indirect mechanisms to shut off the NF-κB-dependent host inflammatory responses. Bordetella spp. exploit the anti-inflammatory cytokine IL-10 to suppress the host immune system. The Bordetella bronchiseptica T3SS effector BopN translocates to the nucleus of the host cell, where it induces the production of IL-10 and downregulates MAPKs. In addition, BopN blocks nuclear translocation of RELA but promotes nuclear translocation of p50 to selectively activate IL-10 expression148. This supports previous observations that B. bronchiseptica uses its T3SS to suppress NF-κB in order to inhibit the induction of innate immune genes such as β-defensins149. The virulence factor LcrV of Yersinia spp. also enhances the production of IL-10 via an association with TLR6 (Ref. 150).
Similarly to viruses and bacteria, parasites have acquired diverse mechanisms to modulate the host innate immune responses controlled by NF-κB. For example, the intracellular parasite Toxoplasma gondii increases the level of phosphorylated IκBα in the infected host through the parasite kinase IKK to activate NF-κB and thereby prolong the survival of host cells151,152.
Concluding remarks
NF-κB plays a vital part in the early stages of the host response against diverse pathogens; thus, during the course of evolution microbial pathogens have collectively acquired an impressive repertoire of molecules that target almost every aspect of the NF-κB signalling pathway. These pathogen-derived countermeasures have been selected to maintain a delicate balance between the activation and inhibition of the NF-κB pathway as a survival strategy, and this supervening control has to be exercised throughout the time that the pathogen lives within the host. In fact, single pathogens frequently deploy multiple strategies to modulate the NF-κB circuitry, but it is still a mystery why individual pathogens often need to express so many seemingly redundant regulators of the same pathway (for example, VACV encodes six known distinct NF-κB regulators, and that number is still growing). It is also clear that diverse microbial pathogens have evolved, in parallel, the ability to manipulate common cellular targets to modulate the NF-κB pathway. In fact, the pathogen-derived effector molecules themselves are remarkably diverse. With the continuing identification of ever more cellular mechanisms that regulate NF-κB, it would not be surprising if at least some successful pathogens were shown to subvert these newly found regulatory mechanisms for their benefit. Furthermore, the identification of new pathogen-derived molecules that target NF-κB will undoubtedly increase our appreciation of the most effective ways to manipulate NF-κB therapeutically in uninfected hosts.
The receptors that trigger NF-κB are activated not only by pathogens but also by various molecules that are produced by the host, and the uncontrolled activation of NF-κB is associated with multiple inflammatory diseases, progressing syndromes of autoimmunity, and human cancers. Although the cause-and-effect relationships of NF-κB activation in cancerous tissues are not completely understood, aberrant activation of kinase pathways that feed into the IKK proteins can constitutively activate NF-κB and contribute to the progression of various malignancies153. It is not accidental that the major chemical and physical carcinogens that have been implicated in the promotion of cancer can also frequently activate NF-κB. Newer drugs that inhibit NF-κB by targeting upstream kinases in the pathway, and the IKK proteins, have shown promise as anticancer therapeutics in preclinical studies. However, it is worth remembering that successful pathogens are still Mother Nature's master drug chemists, and pathogen-derived molecules themselves (or derivatives of these molecules), such as bioactive peptides, may be developed as both anticancer and anti-inflammatory therapeutics in the future. Newer generations of drugs are needed to treat those immune diseases and cancers that depend on NF-κB-mediated inflammatory support. Indeed, the 'right' druggable cellular targets have probably already been identified and targeted for exploitation by nature's successful microbial pathogens.
References
Hoffmann, A. & Baltimore, D. Circuitry of nuclear factor κB signaling. Immunol. Rev. 210, 171–186 (2006).
Vallabhapurapu, S. & Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 (2009).
Hayden, M. S. & Ghosh, S. Shared principles in NF-κB signaling. Cell 132, 344–362 (2008). This article summarizes the basic principles of cellular activation and inhibition of NF-κB signalling.
Finlay, B. B. & McFadden, G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782 (2006).
Bowie, A. G. & Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nature Rev. Immunol. 8, 911–922 (2008).
Ghosh, S. & Hayden, M. S. New regulators of NF-κB in inflammation. Nature Rev. Immunol. 8, 837–848 (2008).
Wan, F. & Lenardo, M. J. The nuclear signaling of NF-κB: current knowledge, new insights, and future perspectives. Cell Res. 20, 24–33 (2010).
Solt, L. A. & May, M. J. The IκB kinase complex: master regulator of NF-κB signaling. Immunol. Res. 42, 3–18 (2008).
Hacker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13 (2006).
Chen, Z. J. Ubiquitin signalling in the NF-κB pathway. Nature Cell Biol. 7, 758–765 (2005).
Pham, A. M. & tenOever, B. R. The IKK kinases: operators of antiviral signaling. Viruses 2, 55–72 (2010).
Peters, R. T. & Maniatis, T. A new family of IKK-related kinases may function as IκB kinase kinases. Biochim. Biophys. Acta 1471, M57–M62 (2001).
Gilmore, T. D. Introduction to NF-κB: players, pathways, perspectives. Oncogene 25, 6680–6684 (2006).
Basak, S. & Hoffmann, A. Crosstalk via the NF-κB signaling system. Cytokine Growth Factor Rev. 19, 187–197 (2008).
Wang, X. W., Tan, N. S., Ho, B. & Ding, J. L. Evidence for the ancient origin of the NF-κB/IκB cascade: its archaic role in pathogen infection and immunity. Proc. Natl Acad. Sci. USA 103, 4204–4209 (2006). This study shows that the NF-κB signalling pathway is evolutionarily conserved and plays a part in pathogen infection and immunity.
Kawai, T. & Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337 (2009).
Tato, C. M. & Hunter, C. A. Host-pathogen interactions: subversion and utilization of the NF-κ B pathway during infection. Infect. Immun. 70, 3311–3317 (2002).
Wang, J. et al. NF-κB RelA subunit is crucial for early IFN-β expression and resistance to RNA virus replication. J. Immunol. 185, 1720–1729 (2010).
Hiscott, J., Nguyen, T. L., Arguello, M., Nakhaei, P. & Paz, S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene 25, 6844–6867 (2006).
de Oliveira, D. E., Ballon, G. & Cesarman, E. NF-κB signaling modulation by EBV and KSHV. Trends Microbiol. 18, 248–257 (2010).
Santoro, M. G., Rossi, A. & Amici, C. NF-κB and virus infection: who controls whom. EMBO J. 22, 2552–2560 (2003).
Mulhern, O., Harrington, B. & Bowie, A. G. Modulation of innate immune signalling pathways by viral proteins. Adv. Exp. Med. Biol. 666, 49–63 (2009).
Rahman, M. M. & McFadden, G. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2, e4 (2006).
Unterholzner, L. & Bowie, A. G. The interplay between viruses and innate immune signaling: recent insights and therapeutic opportunities. Biochem. Pharmacol. 75, 589–602 (2008).
Sugrue, R. J. Interactions between respiratory syncytial virus and the host cell: opportunities for antivirus strategies? Expert Rev. Mol. Med. 8, 1–17 (2006).
Yoboua, F., Martel, A., Duval, A., Mukawera, E. & Grandvaux, N. Respiratory syncytial virus-mediated NF-κB p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKKβ. J. Virol. 84, 7267–7277 (2010).
Choudhary, S., Boldogh, S., Garofalo, R., Jamaluddin, M. & Brasier, A. R. Respiratory syncytial virus influences NF-κB-dependent gene expression through a novel pathway involving MAP3K14/NIK expression and nuclear complex formation with NF-κB2. J. Virol. 79, 8948–8959 (2005).
Kurt-Jones, E. A. et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature Immunol. 1, 398–401 (2000).
Reimers, K., Buchholz, K. & Werchau, H. Respiratory syncytial virus M2–1 protein induces the activation of nuclear factor kappa B. Virology 331, 260–268 (2005).
Demarchi, F., Gutierrez, M. I. & Giacca, M. Human immunodeficiency virus type 1 Tat protein activates transcription factor NF-κB through the cellular interferon-inducible, double-stranded RNA-dependent protein kinase, PKR. J. Virol. 73, 7080–7086 (1999).
Furia, B. et al. Enhancement of nuclear factor-κB acetylation by coactivator p300 and HIV-1 Tat proteins. J. Biol. Chem. 277, 4973–4980 (2002).
Varin, A. et al. Exogenous Nef protein activates NF-κB, AP-1, and c-Jun N-terminal kinase and stimulates HIV transcription in promonocytic cells. Role in AIDS pathogenesis. J. Biol. Chem. 278, 2219–2227 (2003).
Varin, A. et al. Synthetic Vpr protein activates activator protein-1, c-Jun N-terminal kinase, and NF-κB and stimulates HIV-1 transcription in promonocytic cells and primary macrophages. J. Biol. Chem. 280, 42557–42567 (2005).
Higuchi, M. & Fujii, M. Distinct functions of HTLV-1 Tax1 from HTLV-2 Tax2 contribute key roles to viral pathogenesis. Retrovirology 6, 117 (2009).
Sun, S. C. & Yamaoka, S. Activation of NF-κB by HTLV-I and implications for cell transformation. Oncogene 24, 5952–5964 (2005).
Harhaj, E. W. et al. Somatic mutagenesis studies of NF-κB signaling in human T cells: evidence for an essential role of IKKγ in NF-κB activation by T-cell costimulatory signals and HTLV-I Tax protein. Oncogene 19, 1448–1456 (2000).
Wu, X. & Sun, S. C. Retroviral oncoprotein Tax deregulates NF-κB by activating Tak1 and mediating the physical association of Tak1–IKK. EMBO Rep. 8, 510–515 (2007).
Huang, J., Ren, T., Guan, H., Jiang, Y. & Cheng, H. HTLV-1 Tax is a critical lipid raft modulator that hijacks IκB kinases to the microdomains for persistent activation of NF-κB. J. Biol. Chem. 284, 6208–6217 (2009).
Xiao, G. et al. Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J. 20, 6805–6815 (2001).
Wang, J. et al. BFV activates the NF-κB pathway through its transactivator (BTas) to enhance viral transcription. Virology 400, 215–223 (2010).
Gaudreault, E., Fiola, S., Olivier, M. & Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 81, 8016–8024 (2007).
Mosialos, G. et al. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80, 389–399 (1995). This study is the first to demonstrate that EBV LMP1 functions as a constitutively activated TNFR and activates the NF-κB signalling pathway.
Paine, E., Scheinman, R. I., Baldwin, A. S. Jr & Raab-Traub, N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-κB/Rel family proteins. J. Virol. 69, 4572–4576 (1995).
Huen, D. S., Henderson, S. A., Croom-Carter, D. & Rowe, M. The Epstein–Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10, 549–560 (1995).
Wu, L., Nakano, H. & Wu, Z. The C-terminal activating region 2 of the Epstein-Barr virus-encoded latent membrane protein 1 activates NF-κB through TRAF6 and TAK1. J. Biol. Chem. 281, 2162–2169 (2006).
Luftig, M. et al. Epstein–Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKKα-dependent noncanonical NF-κB activation. Proc. Natl Acad. Sci. USA 101, 141–146 (2004). This study demonstrates that EBV LMP1 activates the alternative NF-κB signalling pathway.
Kung, C. P. & Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF- κB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J. Virol. 84, 6605–6614 (2010).
Bagneris, C. et al. Crystal structure of a vFlip-IKKγ complex: insights into viral activation of the IKK signalosome. Mol. Cell 30, 620–631 (2008).
Matta, H. et al. A nuclear role for Kaposi's sarcoma-associated herpesvirus-encoded K13 protein in gene regulation. Oncogene 27, 5243–5253 (2008).
Ye, F. C. et al. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-κB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 82, 4235–4249 (2008).
Punj, V. et al. Kaposi's sarcoma-associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 suppresses CXCR4 expression by upregulating miR-146a. Oncogene 29, 1835–1844 (2010).
Chung, Y. H. et al. STP-C, an oncoprotein of herpesvirus saimiri augments the activation of NF-κB through ubiquitination of TRAF6. J. Biochem. Mol. Biol. 40, 341–348 (2007).
Cho, I. R. et al. Activation of non-canonical NF-κB pathway mediated by STP-A11, an oncoprotein of Herpesvirus saimiri. Virology 359, 37–45 (2007).
de Jong, S. J., Albrecht, J. C., Schmidt, M., Muller-Fleckenstein, I. & Biesinger, B. Activation of noncanonical NF-κB signaling by the oncoprotein Tio. J. Biol. Chem. 285, 16495–16503 (2010).
Dong, X. et al. Murine gamma-herpesvirus 68 hijacks MAVS and IKKβ to initiate lytic replication. PLoS Pathog. 6, e1001001 (2010).
Brown, H. J. et al. NF-κB inhibits gammaherpesvirus lytic replication. J. Virol. 77, 8532–8540 (2003).
Zhang, X., Zhang, H. & Ye, L. Effects of hepatitis B virus X protein on the development of liver cancer. J. Lab. Clin. Med. 147, 58–66 (2006).
Bouchard, M. J. & Schneider, R. J. The enigmatic X gene of hepatitis B virus. J. Virol. 78, 12725–12734 (2004).
Bui-Nguyen, T. M. et al. NF-κB signaling mediates the induction of MTA1 by hepatitis B virus transactivator protein HBx. Oncogene 29, 1179–1189 (2010).
Chang, S., Dolganiuc, A. & Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82, 479–487 (2007).
Rodriguez, C. I. et al. African swine fever virus IAP-like protein induces the activation of nuclear factor κB. J. Virol. 76, 3936–3942 (2002).
Sciortino, M. T. et al. Involvement of HVEM receptor in activation of nuclear factor κB by herpes simplex virus 1 glycoprotein D. Cell. Microbiol. 10, 2297–2311 (2008).
Liu, X., Fitzgerald, K., Kurt-Jones, E., Finberg, R. & Knipe, D. M. Herpesvirus tegument protein activates NF-κB signaling through the TRAF6 adaptor protein. Proc. Natl Acad. Sci. USA 105, 11335–11339 (2008).
Hargett, D., Rice, S. & Bachenheimer, S. L. Herpes simplex virus type 1 ICP27-dependent activation of NF-κB. J. Virol. 80, 10565–10578 (2006).
van Lint, A. L. et al. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-κB signaling. J. Virol. 84, 10802–10811 (2010).
Kim, J. C. et al. HSV-1 ICP27 suppresses NF-κB activity by stabilizing IκBα. FEBS Lett. 582, 2371–2376 (2008).
Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl Acad. Sci. USA 102, 2992–2997 (2005).
Li, X. D., Sun, L., Seth, R. B., Pineda, G. & Chen, Z. J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl Acad. Sci. USA 102, 17717–17722 (2005).
Abe, T. et al. Hepatitis C virus nonstructural protein 5A modulates the Toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81, 8953–8966 (2007).
Zahoor, M. A. et al. Bovine viral diarrhea virus non-structural protein 5A interacts with NIK- and IKKβ-binding protein. J. Gen. Virol. 91, 1939–1948 (2010).
Choi, S. H. et al. Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alpha signaling through effects on cellular IκB kinase. Mol. Cell Biol. 26, 3048–3059 (2006).
Joo, M. et al. Hepatitis C virus core protein suppresses NF-κB activation and cyclooxygenase-2 expression by direct interaction with IκB kinase β. J. Virol. 79, 7648–7657 (2005).
Fang, X. et al. The membrane protein of SARS-CoV suppresses NF-κB activation. J. Med. Virol. 79, 1431–1439 (2007).
Valentine, R. et al. Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-κB pathway in carcinoma cells by inhibiting IKK phosphorylation. Mol. Cancer 9, 1 (2010).
Mohamed, M. R. & McFadden, G. NFκB inhibitors: strategies from poxviruses. Cell Cycle 8, 3125–3132 (2009).This article summarizes the poxvirus modulators of NF-κB-signalling.
Harte, M. T. et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197, 343–351 (2003).
Stack, J. et al. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201, 1007–1018 (2005).
Graham, S. C. et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 4, e1000128 (2008).
DiPerna, G. et al. Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 279, 36570–36578 (2004).
Chen, R. A., Ryzhakov, G., Cooray, S., Randow, F. & Smith, G. L. Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4, e22 (2008).
McCoy, L. E., Fahy, A. S., Chen, R. A. & Smith, G. L. Mutations in modified virus Ankara protein 183 render it a non-functional counterpart of B14, an inhibitor of nuclear factor κB activation. J. Gen. Virol. 91, 2216–2220 (2010).
Meng, X. et al. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J. Virol. 83, 10627–10636 (2009).
Myskiw, C., Arsenio, J., van Bruggen, R., Deschambault, Y. & Cao, J. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-κB, and IRF3 pathways. J. Virol. 83, 6757–6768 (2009).
Murao, L. E. & Shisler, J. L. The MCV MC159 protein inhibits late, but not early, events of TNF-α-induced NF-κB activation. Virology 340, 255–264 (2005).
Challa, S., Woelfel, M., Guildford, M., Moquin, D. & Chan, F. K. Viral cell death inhibitor MC159 enhances innate immunity against vaccinia virus infection. J. Virol. 84, 10467–10476 (2010).
Nichols, D. B. & Shisler, J. L. Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF-κB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J. Virol. 83, 3162–3174 (2009).
Diel, D. G., Delhon, G., Luo, S., Flores, E. F. & Rock, D. L. A novel inhibitor of the NF-κB signaling pathway encoded by the parapoxvirus orf virus. J. Virol. 84, 3962–3973 (2010).
Tait, S. W., Reid, E. B., Greaves, D. R., Wileman, T. E. & Powell, P. P. Mechanism of inactivation of NF-κB by a viral homologue of IκBα. Signal-induced release of IκBα results in binding of the viral homologue to NF-κB. J. Biol. Chem. 275, 34656–34664 (2000). This paper describes the first viral homologue of IκBα that interacts with cellular RELA and suppresses NF-κB activation.
Zaragoza, C. et al. Viral protease cleavage of inhibitor of κBα triggers host cell apoptosis. Proc. Natl Acad. Sci. USA 103, 19051–19056 (2006). This study shows that the coxsackievirus protease directly cleaves IκBα for inhibition of the NF-κB pathway.
Doceul, V. et al. The Npro product of classical swine fever virus interacts with IκBα, the NF-κB inhibitor. J. Gen. Virol. 89, 1881–1889 (2008).
Bour, S., Perrin, C., Akari, H. & Strebel, K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-κB activation by interfering with βTrCP-mediated degradation of IκB. J. Biol. Chem. 276, 15920–15928 (2001).
Akari, H., Bour, S., Kao, S., Adachi, A. & Strebel, K. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κB-dependent expression of antiapoptotic factors. J. Exp. Med. 194, 1299–1311 (2001).
Graff, J. W., Ettayebi, K. & Hardy, M. E. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of β-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5, e1000280 (2009).
Yim, H. C., Li, J. C., Lau, J. S. & Lau, A. S. HIV-1 Tat dysregulation of lipopolysaccharide-induced cytokine responses: microbial interactions in HIV infection. AIDS 23, 1473–1484 (2009).
Neznanov, N. et al. Proteolytic cleavage of the p65-RelA subunit of NF-κB during poliovirus infection. J. Biol. Chem. 280, 24153–24158 (2005).
Chang, S. J. et al. Poxvirus host range protein CP77 contains an F-box-like domain that is necessary to suppress NF-κB activation by tumor necrosis factor alpha but is independent of its host range function. J. Virol. 83, 4140–4152 (2009).
Mohamed, M. R. et al. Proteomic screening of variola virus reveals a unique NF-κB inhibitor that is highly conserved among pathogenic orthopoxviruses. Proc. Natl Acad. Sci. USA 106, 9045–9050 (2009). This paper describes a class of poxvirus immunomodulatory proteins that inhibit the NF-κB pathway by targeting p105.
Rahman, M. M., Mohamed, M. R., Kim, M., Smallwood, S. & McFadden, G. Co-regulation of NF-κB and inflammasome-mediated inflammatory responses by myxoma virus pyrin domain-containing protein M013. PLoS Pathog. 5, e1000635 (2009).
Taylor, S. L., Frias-Staheli, N., Garcia-Sastre, A. & Schmaljohn, C. S. Hantaan virus nucleocapsid protein binds to importin α proteins and inhibits tumor necrosis factor α-induced activation of nuclear factor κB. J. Virol. 83, 1271–1279 (2009).
Jiao, J., Guan, H., Lippa, A. M. & Ricciardi, R. P. The N terminus of adenovirus type 12 E1A inhibits major histocompatibility complex class I expression by preventing phosphorylation of NF-κB p65 Ser276 through direct binding. J. Virol. 84, 7668–7674 (2010).
Guan, H., Jiao, J. & Ricciardi, R. P. Tumorigenic adenovirus type 12 E1A inhibits phosphorylation of NF-κB by PKAc, causing loss of DNA binding and transactivation. J. Virol. 82, 40–48 (2008).
Taylor, R. T. & Bresnahan, W. A. Human cytomegalovirus IE86 attenuates virus- and tumor necrosis factor α-induced NFκB-dependent gene expression. J. Virol. 80, 10763–10771 (2006).
Jarvis, M. A. et al. Human cytomegalovirus attenuates interleukin-1β and tumor necrosis factor α proinflammatory signaling by inhibition of NF-κB activation. J. Virol. 80, 5588–5598 (2006).
Montag, C., Wagner, J., Gruska, I. & Hagemeier, C. Human cytomegalovirus blocks tumor necrosis factor α- and interleukin-1β-mediated NF-κB signaling. J. Virol. 80, 11686–11698 (2006).
Nachtwey, J. & Spencer, J. V. HCMV IL-10 suppresses cytokine expression in monocytes through inhibition of nuclear factor-κB. Viral Immunol. 21, 477–482 (2008).
Hansberger, M. W. et al. IκB kinase subunits α and γ are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J. Virol. 81, 1360–1371 (2007).
Clarke, P., Debiasi, R. L., Meintzer, S. M., Robinson, B. A. & Tyler, K. L. Inhibition of NF-κB activity and cFLIP expression contribute to viral-induced apoptosis. Apoptosis 10, 513–524 (2005).
Wilson, J. R., de Sessions, P. F., Leon, M. A. & Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 82, 8262–8271 (2008).
Lei, X. et al. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nature Cell Biol. 12, 193–199 (2010).
Bhavsar, A. P., Guttman, J. A. & Finlay, B. B. Manipulation of host-cell pathways by bacterial pathogens. Nature 449, 827–834 (2007).
Naumann, M. Control of the NF-κB inhibitor IκBα in pathogen infection. Biochem. Soc. Trans. 35, 267–269 (2007).
Kvitko, B. H. et al. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5, e1000388 (2009).
Tseng, T. T., Tyler, B. M. & Setubal, J. C. Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 9 (Suppl. 1), S2 (2009).
Viboud, G. I. & Bliska, J. B. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89 (2005).
Zhou, H. et al. Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-κB activation. J. Exp. Med. 202, 1327–1332 (2005).
Sweet, C. R., Conlon, J., Golenbock, D. T., Goguen, J. & Silverman, N. YopJ targets TRAF proteins to inhibit TLR-mediated NF-κB, MAPK and IRF3 signal transduction. Cell. Microbiol. 9, 2700–2715 (2007).
Mukherjee, S. et al. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 (2006). This study shows that YopJ acts as an acetyltransferase for acetylation of IKKα and IKKβ and blocks activation of the IKK complex.
Mittal, R., Peak-Chew, S. Y. & McMahon, H. T. Acetylation of MEK2 and IκB kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl Acad. Sci. USA 103, 18574–18579 (2006).
Fehr, D. et al. AopP, a type III effector protein of Aeromonas salmonicida, inhibits the NF-κB signalling pathway. Microbiology 152, 2809–2818 (2006).
Brummelkamp, T. R., Nijman, S. M., Dirac, A. M. & Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 424, 797–801 (2003).
Wertz, I. E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694–699 (2004).
Ye, Z., Petrof, E. O., Boone, D., Claud, E. C. & Sun, J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 171, 882–892 (2007).
Collier-Hyams, L. S. et al. Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF-κB pathway. J. Immunol. 169, 2846–2850 (2002).
Rytkonen, A. et al. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl Acad. Sci. USA 104, 3502–3507 (2007).
Le Negrate, G. et al. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-κB, suppresses IκBα ubiquitination and modulates innate immune responses. J. Immunol. 180, 5045–5056 (2008).
Kim, D. W. et al. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl Acad. Sci. USA 102, 14046–14051 (2005). This work finds that the S. flexneri T3SS effector protein OspG blocks degradation of phosphorylated IκBα by targeting the cellular ubiquitin machinery.
Gao, X. et al. Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 5, e1000708 (2009). This article reports the identification of nleH genes (from EHEC) that regulate the transcriptional function of NF-κB in the nucleus by interaction with RPS3.
Nobe, R. et al. Enterohaemorrhagic Escherichia coli serogroup O111 inhibits NF-κB-dependent innate responses in a manner independent of a type III secreted OspG orthologue. Microbiology 155, 3214–3225 (2009).
Malladi, V., Puthenedam, M., Williams, P. H. & Balakrishnan, A. Enteropathogenic Escherichia coli outer membrane proteins induce iNOS by activation of NF-κB and MAP kinases. Inflammation 28, 345–353 (2004).
Nadler, C. et al. The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog. 6, e1000743 (2010).
Newton, H. J. et al. The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6, e1000898 (2010).
Ashida, H. et al. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nature Cell Biol. 12, 66–73 (2010).
Selvaraj, S. K. & Prasadarao, N. V. Escherichia coli K1 inhibits proinflammatory cytokine induction in monocytes by preventing NF-κB activation. J. Leukoc. Biol. 78, 544–554 (2005).
Kravchenko, V. V. et al. Modulation of gene expression via disruption of NF-κB signaling by a bacterial small molecule. Science 321, 259–263 (2008).
Bhattacharjee, R. N. et al. VP1686, a Vibrio type III secretion protein, induces Toll-like receptor-independent apoptosis in macrophage through NF-κB inhibition. J. Biol. Chem. 281, 36897–36904 (2006).
Betts, H. J., Wolf, K. & Fields, K. A. Effector protein modulation of host cells: examples in the Chlamydia spp. arsenal. Curr. Opin. Microbiol. 12, 81–87 (2009). This paper describes how the intracellular bacterial pathogens of the genus Chlamydia deploy multiple pathogen-encoded proteins for NF-κB regulation through multiple mechanisms.
Lad, S. P. et al. Cleavage of p65/RelA of the NF-κB pathway by Chlamydia. Proc. Natl Acad. Sci. USA 104, 2933–2938 (2007).
Le Negrate, G. et al. ChlaDub1 of Chlamydia trachomatis suppresses NF-κB activation and inhibits IκBα ubiquitination and degradation. Cell. Microbiol. 10, 1879–1892 (2008).
Wolf, K., Plano, G. V. & Fields, K. A. A protein secreted by the respiratory pathogen Chlamydia pneumoniae impairs IL-17 signaling via interaction with human Act1. Cell. Microbiol. 11, 769–779 (2009).
Ge, J. et al. A Legionella type IV effector activates the NF-κB pathway by phosphorylating the IκB family of inhibitors. Proc. Natl Acad. Sci. USA 106, 13725–13730 (2009). This study shows that the bacterial pathogen Legionella pneumophila activates NF-κB signalling by encoding a Ser/Thr kinase that directly phosphorylates IκBα.
Clifton, D. R., Rydkina, E., Freeman, R. S. & Sahni, S. K. NF-κB activation during Rickettsia rickettsii infection of endothelial cells involves the activation of catalytic IκB kinases IKKα and IKKβ and phosphorylation-proteolysis of the inhibitor protein IκBα. Infect. Immun. 73, 155–165 (2005).
Abramson, T., Kedem, H. & Relman, D. A. Modulation of the NF-κB pathway by Bordetella pertussis filamentous hemagglutinin. PLoS ONE 3, e3825 (2008).
Carneiro, L. A. et al. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5, 123–136 (2009).
Wells, J. M., Loonen, L. M. & Karczewski, J. M. The role of innate signaling in the homeostasis of tolerance and immunity in the intestine. Int. J. Med. Microbiol. 300, 41–48 (2010).
Iyer, C. et al. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-κB and MAPK signalling. Cell. Microbiol. 10, 1442–1452 (2008).This investigation finds that probiotics downregulate NF-κB signalling pathways, enabling them to form symbiotic relationships with the host.
van Baarlen, P. et al. Differential NF kappa B pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc. Natl Acad. Sci. USA 106, 2371–2376 (2009).
Guarino, A., Lo Vecchio, A. & Canani, R. B. Probiotics as prevention and treatment for diarrhea. Curr. Opin. Gastroenterol. 25, 18–23 (2009).
Nagamatsu, K. et al. Bordetella evades the host immune system by inducing IL-10 through a type III effector, BopN. J. Exp. Med. 206, 3073–3088 (2009).
Legarda, D., Klein-Patel, M. E., Yim, S., Yuk, M. H. & Diamond, G. Suppression of NF-κB-mediated β-defensin gene expression in the mammalian airway by the Bordetella type III secretion system. Cell. Microbiol. 7, 489–497 (2005).
Depaolo, R. W. et al. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4, 350–361 (2008).
Molestina, R. E. & Sinai, A. P. Detection of a novel parasite kinase activity at the Toxoplasma gondii parasitophorous vacuole membrane capable of phosphorylating host IκBα. Cell. Microbiol. 7, 351–362 (2005).
Molestina, R. E. & Sinai, A. P. Host and parasite-derived IKK activities direct distinct temporal phases of NF-κB activation and target gene expression following Toxoplasma gondii infection. J. Cell Sci. 118, 5785–5796 (2005).
Van Waes, C. Nuclear factor-κB in development, prevention, and therapy of cancer. Clin. Cancer Res. 13, 1076–1082 (2007).
Song, Y. J. & Kang, M. S. Roles of TRAF2 and TRAF3 in Epstein-Barr virus latent membrane protein 1-induced alternative NF-κB activation. Virus Genes 41, 174–180 (2010).
Brinkmann, M. M. et al. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77, 9346–9358 (2003).
Friedman, J. M. & Horwitz, M. S. Inhibition of tumor necrosis factor α-induced NF-κB activation by the adenovirus E3–104/14.5K complex. J. Virol. 76, 5515–5521 (2002).
Unterstab, G. et al. Viral targeting of the interferon-β-inducing Traf family member-associated NF-κB activator (TANK)-binding kinase-1. Proc. Natl Acad. Sci. USA 102, 13640–13645 (2005).
Mohamed, M. R. et al. Cowpox virus expresses a novel ankyrin repeat NF-κB inhibitor that controls inflammatory cell influx into virus-infected tissues and is critical for virus pathogenesis. J. Virol. 83, 9223–9236 (2009).
Park, K. J. et al. Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 278, 30711–30718 (2003).
Mack, C., Sickmann, A., Lembo, D. & Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl Acad. Sci. USA 105, 3094–3099 (2008).
Spitkovsky, D., Hehner, S. P., Hofmann, T. G., Moller, A. & Schmitz, M. L. The human papillomavirus oncoprotein E7 attenuates NF-κB activation by targeting the IκB kinase complex. J. Biol. Chem. 277, 25576–25582 (2002).
Nichols, D. B. & Shisler, J. L. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor α-induced NF-κB activation via inhibition of Iκ kinase complex formation. J. Virol. 80, 578–586 (2006).
Shisler, J. L. & Jin, X. L. The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J. Virol. 78, 3553–3560 (2004).
Gedey, R., Jin, X. L., Hinthong, O. & Shisler, J. L. Poxviral regulation of the host NF-κB response: the vaccinia virus M2L protein inhibits induction of NF-κB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 80, 8676–8685 (2006).
Jones, J. O. & Arvin, A. M. Inhibition of the NF-κB pathway by varicella-zoster virus in vitro and in human epidermal cells in vivo. J. Virol. 80, 5113–5124 (2006).
Heussler, V. T. et al. Hijacking of host cell IKK signalosomes by the transforming parasite Theileria. Science 298, 1033–1036 (2002).This work demonstrates that the intracellular protozoan parasites of the genus Theileria constitutively activate the NF-κB pathway by modulating the signalling of the cellular IKK complex.
Trosky, J. E. et al. Inhibition of MAPK signaling pathways by VopA from Vibrio parahaemolyticus. J. Biol. Chem. 279, 51953–51957 (2004).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rahman, M., McFadden, G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol 9, 291–306 (2011). https://doi.org/10.1038/nrmicro2539
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2539
This article is cited by
-
Disparate macrophage responses are linked to infection outcome of Hantan virus in humans or rodents
Nature Communications (2024)
-
A comprehensive model for the biochemistry of ageing, senescence and longevity
Biogerontology (2024)
-
The NF-ĸB p50 subunit generated by KPC1-mediated ubiquitination and limited proteasomal processing, suppresses tumor growth
Cancer Cell International (2023)
-
TLR2-mediated NF-κB signaling pathway is involved in PPV1-induced apoptosis in PK-15 cells
Veterinary Research Communications (2023)
-
Oral streptococci subvert the host innate immune response through hydrogen peroxide
Scientific Reports (2022)
