
Key Points
-
Noroviruses are a major cause of gastroenteritis, and there are currently no vaccines or antiviral treatments available to treat or prevent the >260 million gastroenteritis cases that are reported globally each year. Noroviruses have proven difficult to work with in the laboratory owing to the lack of cell culture systems and animal models, and therefore little is known about the pathogenesis caused by this virus, which has hampered the development of efficacious therapeutics.
-
The norovirus family contains two genogroups (GI and GII) that are most commonly associated with enteric disease in humans, and these genogroups contain more than 25 different genotypes that account for most human norovirus cases. However, outbreaks caused by the GII.4 genotype occur much more frequently than those caused by other genotypes in the GII genogroup, and GI outbreaks occur even less frequently. Although the majority of norovirus outbreaks are caused by the GII.4 genotype, the molecular and biological factors that regulate this disease burden are only partially understood.
-
The GII.4 genotype seems to operate in a similar fashion as influenza virus, whereby evolution of novel immune escape variants allows the virus to escape the predominant memory immune response. By contrast, the prototypic GI.1 noroviruses have remained relatively static over the same time period, evolving variants with identical histo-blood group antigen binding capabilities and similar antigenic properties. The molecular mechanisms governing differential evolution patterns remain a key mystery in the norovirus field.
-
Immunity against noroviruses has been difficult to assess owing to the complex effects of host pre-exposure histories and differential host susceptibility, which is correlated with blood group and secretor status. However, recent work has suggested that the GI and GII genogroups may use different mechanisms to escape immunological memory and that this is perhaps directly related to the plasticity of and complex evolutionary-related sequence information encoded in the P2 subdomain of the capsid protein.
-
The GII genogroup contains more amino acid sequence in the P2 subdomain, which may allow increased capsid plasticity and a tolerance for more amino acid variation or insertions and deletions. This would provide a larger repertoire of sequence targets for natural selection and adaptation to complex environmental selection processes, like herd immunity. By contrast, the GI genogroup contains less sequence information with more conserved, surface-exposed residues that are probably recognized by homologous antibodies as well as antibodies generated against heterologous GI strains. Thus, complex patterns of GI pre-exposure history, antibody cross reactivity and original antigenic sin may facilitate secondary infections of GI strains, whereas antigenic drift and receptor switching allow GII noroviruses, especially GII.4 viruses, to persist in human populations.
Abstract
Noroviruses are the most common cause of food-borne gastroenteritis worldwide, and explosive outbreaks frequently occur in community settings, where the virus can immobilize large numbers of infected individuals for 24–48 hours, making the development of effective vaccines and antiviral therapies a priority. However, several challenges have hampered therapeutic design, including: the limitations of cell culture and small-animal model systems; the complex effects of host pre-exposure histories; differential host susceptibility, which is correlated with blood group and secretor status; and the evolution of novel immune escape variants. In this Review, we discuss the molecular and structural mechanisms that facilitate the persistence of noroviruses in human populations.
Similar content being viewed by others

Main
Noroviruses are the primary etiological agent responsible for most cases of viral gastroenteritis worldwide1, making these viruses the most common cause of sporadic diarrhoea in community settings and a major burden to the military, the cruise ship industry, university campuses, hospitals and retirement communities. In the United States, the CDC estimates that there are ∼23 million annual norovirus infections2, many among the elderly, with outbreaks commonly occurring in retirement communities3,4. In fact, norovirus-associated mortalities are not uncommon in elderly populations, although severe gastroenteritis lasting for 12–48 hours is the predominant disease outcome in individuals of all ages5.
Early seminal studies identified the norovirus particle using immuno-electron microscopy and human challenge studies to observe and verify noroviruses in faecal samples derived from children infected during an epidemic outbreak in Norwalk, Ohio, USA6,7. Subsequently, the Norwalk virus genome was cloned and sequenced8.
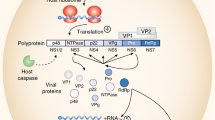
Noroviruses belong to the family Caliciviridae and are small, non-enveloped, icosahedral viruses with a diameter of ∼38 nm. The viral genome is a ∼7.5 kb, positive-sense, single-stranded RNA containing three open reading frames that encode both the structural and non-structural proteins (Fig. 1a). The capsid protein, viral protein 1 (VP1), is the most important component of the viral capsid, with 180 copies found in the intact viral particle, whereas VP2, a minor structural protein, is incorporated into the capsid in low copy number (Fig.1).

a| The norovirus genome is composed of three open reading frames (ORFs). ORF1 (∼5 kb) is located in the first two-thirds of the genome and encodes a ∼200 kDa polyprotein that is auto-processed by a virally encoded 3C-like protease (3C) to yield the non-structural replicase proteins that are essential for viral replication89. The resultant proteins are: p48, an amino-terminal protein of unknown function (∼48 kDa); nucleoside triphosphatase (NTP), a 2C-like protein; p22, a 22 kDa 3A-like protein; viral genome-linked protein (VPG), a protein that is covalently linked to the 5′ end of the genome; and RNA-directed RNA polymerase (RdRp), a 3D-like protein. ORF2 is 1.8kb in length and encodes the 57 kDa major structural capsid protein, viral protein 1 (VP1). VP1 is divided into two domains, the shell domain (yellow) and the protruding domain, which is further divided into two subdomains known as P1 (blue) and P2 (red). ORF3 is ∼0.6 kb in length and encodes a 22 kDa minor basic structural protein, VP2 (Ref. 89). b | The structure of the VP1 monomer is shown, with protein domains coloured as for part a. c | Two capsid protein monomers form the A–B dimer (indicated with the A monomer in lighter shades and the B monomer in darker shades), which allows the P2 domain to protrude from the viral particle. d |The virus-like particle is formed of 180 monomers of the capsid protein that assemble through different dimers. The A–B dimer, shown in colour, extends away from the capsid and provides the receptor-binding region and the sites of antigenic variation. In the virus particle, VP2 is incorporated in low copy number. Structural models were generated and pictures were rendered using MacPyMOL (Delano Scientific LLC, Palo Alto, California, USA).
Norovirus research has been hindered by the lack of of small-animal models and a reliable cell culture system. In response, recombinant expression systems have been developed that allow for the formation of virus-like particles (VLPs), which morphologically and antigenically recapitulate the native norovirus particle (Fig. 1d) and contain strain-specific and group-specific immunogenic determinants9,10,11. Two recombinant expression systems, the baculovirus replicon system9 and the Venezuelan equine encephalitis virus replicon system11, have been well characterized, and both generate copious amounts of norovirus capsid protein that spontaneously self-assembles into VLPs (Box 1). VLP preparations have provided the foundation for most studies into the carbohydrate (CHO) ligand-binding specificities, capsid structures and antigenic relationships of noroviruses and the development of candidate vaccine strains.
Norovirus protective immunity has been a hotly debated topic, with conflicting reports in the literature, some of which suggest that norovirus-induced immunity is strain specific and limited to a short-term immune response, whereas others show possible long-term protection from reinfection12,13; however, these studies have been limited in sample size, and volunteers were probably challenged with a viral quasispecies, rather than a single genotype. These conflicting results have confounded vaccine study design and show that norovirus immunity is more complex than previously appreciated. In this Review article, we discuss recent findings that shed light on the molecular mechanisms governing norovirus capsid evolution, structure and receptor recognition as a function of immune system-driven antigenic drift, and we speculate on how capsid plasticity may have facilitated the broader emergence of particular genotypes and genogroups at the expense of others.
Norovirus phylogeny
The genus Norovirus is composed of more than 40 diverse virus strains divided into 5 genogroups on the basis of sequence similarity14,15,16,17, designated GI–GV, with the GI and GII genogroups being the most important for human infection18. Each genogroup is further divided into genotypes on the basis of pairwise distribution19, with GI containing 8 distinct genotypes and GII at least 17 different genotypes19 (Fig. 2). The genogroup II, genotype 4 noroviruses, designated GII.4, are currently responsible for 70–80% of norovirus outbreaks worldwide, and the Norwalk virus, designated GI.1-NV, is the prototypical norovirus strain.

A Bayesian phylogenetic tree of representative norovirus capsid protein sequences from all major genogroups and genotypes. Labels indicate the GI and GII genogroups that predominantly infect humans, and red brackets represent genotypes that seem to be evolving. The tree was generated with MRBAYES90, using sapovirus Nongkhai 50 to root the tree.
Strains in a genotype have 69–97% nucleotide similarity at the genomic level, whereas strains of different genogroups are more distant, having 51–56% genomic nucleotide similarity20. Furthermore, the structural proteins are even more variable, as strains of one genotype generally differ by up to 40% in capsid amino acid sequence, whereas strains of different genogroups usually differ by more than 50%21.
Most norovirus challenge studies have assumed that individual genotypes represent strains with similar phenotypes, but recent evidence provided by our laboratory and others indicates that evolution in some genotypes is sufficient to generate mutant clusters that have new ligand-binding characteristics and antigenic properties22. In fact, evolution in the GII.4 genotype results in differential receptor binding and novel antigenic features, suggesting that the GII.4 noroviruses are evolving over time, with escape mutants being periodically selected for by herd immunity22,23,24,25. The evolution in this genotype seems to be epochal, as long periods of stasis were followed by bursts of evolution (Fig. 2). In addition, similar, albeit smaller, phylogenetics-based studies of the GII.2 and GII.3 genotypes have suggested analogous patterns of evolution for these genotypes26,27, with evolved clusters that may contain novel innovations that alter viral structure and function. More full-length capsid sequences and further work are necessary to determine if these genotypes are evolving to escape herd immunity.
By contrast, work conducted in our laboratory suggests that the GI genogroup of noroviruses has undergone limited evolution with little phenotypic innovation. Change in the GI noroviruses may be restricted by purifying selection in the capsid protein, whereby structural domains necessary for function are unable to tolerate enough mutation to allow broad escape from immunity. This is dramatically shown in the phylogenetic tree, in which the GI genotypes are less distant from one another compared with the GII genotypes (Fig. 2). Future crystallography studies comparing the structures of multiple GI and GII capsid proteins along with mutagenesis experiments and studies of carbohydrate ligand-binding characteristics should help to elucidate the differences in the structural constraints between these two genogroups, perhaps providing insights into the mechanisms that drove the discordant evolution patterns that have been observed between the broader genogroups and genotypes over the past 40 years.
Histo-blood group antigen receptors
Several studies have indicated that histo-blood group antigens (HBGA) function as receptors or co-receptors for a productive norovirus infection12,28,29,30, although no direct evidence has shown that viral binding to HBGAs mediates entry. However, there is a correlation between polymorphic expression of HBGA and human susceptibility to norovirus infection12. Even early studies with GI.1-NV found that some individuals could not be infected with GI.1-NV on challenge and these resistant individuals typically clustered in families, suggesting that a highly penetrant host susceptibility allele was influencing disease outcomes. It was further proposed that the presence of pre-existing antibody did not correlate with protection31,32.
More recent studies have shown that individuals of blood group O are more susceptible to GI.1-NV infection than individuals of other blood groups29, and it was found that GI.1-NV VLPs bound to gastroduodenal epithelial cells from secretor-positive individuals, whereas VLPs did not bind to the cells of secretor-negative individuals33. The fucosyltransferase 2 gene (FUT2), carried by secretor-positive individuals, encodes a protein that is responsible for generating the H1 and H3 antigens from their respective disaccharide precursors, and these antigens are known to be present on mucosal surfaces and have been shown to bind GI.1-NV VLPs33. Direct evidence of the association between GI.1-NV susceptibility and HBGA expression was shown in a large-scale human challenge study in which secretor-negative individuals were found to be resistant to infection34. Additional studies with other norovirus strains have revealed that susceptibility to infection with other strains is more complicated than susceptibility to GI.1-NV. Although numbers were limited, a study with the Snow Mountain virus of the GII.2 group found that blood group and secretor status did not strongly correlate with susceptibility to infection35. A different study showed that secretor-negative individuals had substantially lower antibody titres to GII.4 norovirus strains than secretor-positive individuals, suggesting that non-secretors are less likely to become infected with the predominant circulating norovirus strains than secretors; however, blood group was irrelevant to infection status36. In other reports, individuals with blood group B had a lower incidence of infection with GI noroviruses than individuals of other blood groups12,30.
These observations suggest that individual norovirus strains may infect only a subset of the human population, but diverse HBGA-binding affinities in noroviruses of the GI and GII genogroups may collectively allow nearly all individuals to be susceptible to one or more strains. Future human challenge studies using more GI and GII prototype strains are needed to discern the precise susceptibility profile for each genotype and strain.
Binding differences of histo-blood group antigens. Distinct norovirus strains have highly variable HBGA-binding patterns. The carbohydrate-binding characteristics of various norovirus VLPs (Table1) have shown that genetically related noroviruses share binding patterns, indicating that the evolution of virus strains may be influenced by HBGA binding. However, specific binding profiles are not genotype or genogroup exclusive37, and in the case of the GII.4 viruses single amino acid replacements seem to drastically alter the binding capacity of the VLP (Table1).
Noroviruses have been categorized into two binding-profile groups: those that bind A or B and H epitopes and those that bind Lewis or H epitopes. Two recent reports have demonstrated that GII.4 noroviruses associated with the Farmington Hills outbreak of 2000–2002 bind both groups22,38. A third group should be noted for which no binding occurs to available synthetic HBGAs, such as the GII.4.2004, GII.4.2005, VA115 (which is a GI.3b virus) and mouse norovirus (MNV1)(which is of the GV genogroup) strains, suggesting that alternative viral receptors may exist or that the appropriate CHO variations are not present in saliva or in synthetically available CHOs. Several factors associated with HBGA regulation and expression need to be further characterized if we are to fully elucidate and appreciate the role of these putative receptors in norovirus pathogenesis. It is known that HBGA types are differentially expressed according to tissue type, such that type 1 core structures are found on surface epithelia of endodermal tissues, whereas type 2 core structures are typically found at the glandular level in ectodermic and mesodermic tissues39. A recent study demonstrated that different norovirus genotypes may specifically recognize type 1 and type 2 CHOs, indicating that different noroviruses may target different tissues39. Furthermore, GII (but, to date, not GI) noroviruses have been shown to bind sialylated glycoconjugates, suggesting that additional modifications to and variations of HBGAs may provide additional binding targets for norovirus40. Studies designed to examine these issues will provide important insight into the complex interaction between the noroviruses and their ligands and will elucidate the influence that these glycoconjugates exert on disease outcomes following norovirus infection.
Capsid structure
Structural analysis of the GI.1-NV VLP capsid protein showed that the capsid is formed of 180 capsid molecules that are organized into 90 dimers with a T =3 icosahedral symmetry41 (where T is triangulation number), using two distinct dimer types to form the higher-order structure42. Both dimer types are composed of identical monomers but have different overall orientations, such that the C–C dimers form protrusions at the icosahedral twofold axes, whereas the A–B dimers form protrusions around the fivefold axes (Fig. 1c,d). Analysis of the capsid monomer indicated that it is composed of two distinct domains, the shell (S) domain and the protruding (P) domain, which are linked by a flexible hinge42 (Fig. 1a,b). The S domain, composed of the first 225 residues of the capsid protein, forms the structural core of the intact viral capsid, and the P domain extends from the surface43. The P domain is further divided into two subdomains, P1 and the hypervariable P2, which protrudes furthest from the capsid shell and contains the putative receptor-binding sites42,44,45,46,47,48. A second high-resolution GI.1 virus structure was recently published showing the protruding domain of GI.1-NV in complex with synthetic A-type and H-type HBGAs. Even though there were differences in the CHO moieties of these two HBGAs, both interacted with the same surface-exposed binding site found in the P2 subdomain of the viral capsid protein44,45. This binding site was highly conserved in the GI genogroup and was verified by mutational analysis, which found that specific amino acids in this epitope are essential to binding45.
In addition, the protruding domain of norovirus strain VA387, a GII.4 virus, has been reported in complex with A-type and B-type HBGAs. Structurally, the GI.1 and GII.4 capsid structures are similar in the S domain and most of the P1 subdomain, and the predominant differences occur in the P2 subdomain. In general, the P2 subdomain of GI.1 viruses contains fewer amino acids and is smaller than that of GII.4 viruses, which contains substantially more loops in the P2 region. Moreover, the HBGA-binding regions were completely different between the two14. The GII.4 genotype P domain structure revealed that residues Thr344, Arg345, Asp374 and Gly442 form a hydrogen-bonding network with the α-fucose group of the A trisaccharide, whereas a second interaction site (at residues 390–395) was predicted to stabilize binding and enhance ligand affinity by weaker, long-distance interactions with the β-galactose ring of the trisaccharide14. By contrast, in the GI.1 virus structure, the β-N-acetylgalactosamine moiety of the A trisaccharide was shown to interact with residues Ser377, Asp327, His329 and Ser380 (Refs 44, 45). Not only were these residues different than those reported for the GII.4 virus interaction, but the mode of binding was also distinct, with the β-N-acetylgalactosamine moiety contributing more to the interaction than the α-fucose moiety in the GII.4 virus structure. This observation showed that VLPs of different genogroups engage the same HBGAs through unique modes of binding, and it also highlights the crucial need for more crystal structures of different norovirus P domains, including time-ordered variants, from different genogroups and genotypes in complex with different HBGAs.
In addition to the strain-specific interactions, long-distance interactions between residues that are proximal to those interacting with the HBGA are likely to be necessary for the stabilization of virus–receptor interactions. Replacement of a Gly with an Asp at position 395 of the GII.4.1987 capsid was sufficient to alter the HBGAs that the mutant VLP bound, even though residue 395 does not directly interact with the CHO ligand22. Moreover, synthetic HBGAs used in the crystallography studies may lack important in vivo modifications to the CHO structures (in vivo molecules will probably be larger and contain modified hydroxyl groups), and these structures may therefore not provide accurate assessments of HBGA binding. Consequently, VLP structures bound to more complex HBGA moieties may be crucial to fully disclose the interaction sites between VLPs and CHOs.
The evolutionary impact on capsid structure
Interestingly, sequence analysis has shown that the GI genotype is limited to 37% variation at the amino acid level, with the primary variation occurring as insertions or deletions in different genotypes. Analysis of the published crystal structures of GI.1-NV and of homology models generated for the different GI capsids found that much of the HBGA-binding pocket remains intact. Residues Ser377, Asp327, His329 and Ser380 were strictly conserved, although inserts between these residues defined the GI.2, GI.3 and GI.4 models (Fig. 3), suggesting that there has been a subtle remodelling of the CHO-binding interfaces between these genotypes, which may account for the binding differences that have been noted between these VLPs (Table 1). Structural variation in each genotype was limited and occurred mostly on the surface, in the P2 domain but away from the binding site (Fig. 3).

The GI genogroup is highlighted in blue, and the GII genogroup is highlighted in green. The P domain dimer structures show the predominant genotypes that have infected humans over the past decade in the United States, recognizing that this may vary elsewhere, globally. Fuschia represents the histo-blood group antigen-binding sites and yellow indicates variation in the P2 subdomains. Structural models were generated using the program Modeller91, and pictures were generated using MacPyMOL (Delano Scientific LLC, Palo Alto, California, USA).
Superimposition of the different GI monomer models onto the GI.1-NV structure showed that insertions or deletions alter the overall structure in each case but do not seem to alter the binding pocket or a conserved structural motif found on the exterior of the GI capsid distal from the binding site (Fig. 4A). This conserved motif occurs on the side of the capsid, opposite the dimer-interface site, and may be an immunogenic epitope that allows antibodies generated against one GI norovirus to cross-react with GI noroviruses of different genotypes. Moreover, the structural models predict that the conserved motif and CHO binding may be functionally linked and structurally constrained.

A | Variation in the GI genogroup. The crystal structure of the P domain of the genogroup I, genotype 1 Norwalk virus (GI.1-NV) was used to generate representative models of all eight GI genotypes. P domain chains are in blue, the histo-blood group antigen (HBGA)-binding sites are in magenta, a conserved structural domain is shown in red and all additional GI models are in black mesh. A a | The annotated GI.1-NV P domain structure. A b | The GI.1-NV P domain monomer with additional GI monomer models superimposed on the structure. A c | The GI.1-NV P domain dimer with additional GI dimer models superimposed on the structure. Note that the binding pocket and conserved domain are mostly preserved. B | Variation in the GII genogroup. The crystal structure of the P domain of GII.4 norovirus VA387 was used to generate representative models of all 17 GII P domains. GII.4 P domain chains are in cyan, the HBGA-binding sites are orange and all additional GII P domain models are in purple mesh. B a | The annotated GII.4 VA387 P domain structure. B b | The GII.4 VA387 P domain monomer with additional GII monomer models superimposed. B c | The GII.4 VA387 P domain dimer with additional GII P domain dimer models superimposed. Note that the binding pockets are structurally different in the GII models. C | Superimposing the GII and GI structural space. Ca | Superimposition of the GII.4 VA387 P domain dimer structure (shown in mesh) onto the GI. 1-NV P domain dimer structure indicates that GII.4 VA387 occupies more structural space than GI.1-NV (the colours of domains are as for parts A and B). Cb | Superimposing the GII monomer structure, indicated by the purple mesh, onto the GI monomer structure, shown in black, indicates that the GII genogroup occupies substantially more space. Cc | Superimposing the GII dimer structure, indicated by the purple mesh, onto the GI dimer structure, shown in black, indicates that the GII genogroup occupies more overall structural space. The additional structural space may allow the GII genogroup more structural flexibility, so that it could hypothetically tolerate more mutations while preserving its capacity to bind a differential receptor repertoire. Structural models were generated using the program Modeller91 and pictures were generated using MacPyMOL (Delano Scientific LLC, Palo Alto, California, USA).
By contrast, modelling the structural differences of the GII monomers showed that insertions or deletions are positioned in locations that allowed substantial remodelling of the capsid surface, such that conservation of the HBGA-binding pocket and a flanking conserved, GI-equivalent, surface-exposed structural motif does not occur (Fig. 4b). Comparing the three-dimensional space occupied by all of the different monomer models of the GI (n=8) and GII (n=17) genogroups indicated that the GI genogroup seems to have limited structural plasticity, as the structural space occupied by this genogroup is limited compared with the space occupied by the GII genogroup (Fig. 4c). This suggests that the GI genogroup may be trapped in a genetic bottleneck, such that limited tolerance of structural variation in the capsid results in a limited subset of changes, preventing the evolution of the large-scale innovations that are necessary to generate and escape from herd immunity. By contrast, the GII genogroup has a larger repertoire of sequence space and flexibility, suggesting that it has evolved the ability to function as a capsid protein in the presence of much greater structural variation (Fig. 4c).
Theoretically, the immune system selects for variation in the distal regions of P2, as well as in some surface-exposed P1 residues, that allows for escape of immunity and amplification of new, immune-resistant strains (Fig. 1d). These strains probably contain novel antigenic epitopes and innovative structural changes to the capsid protein49, which together may contribute to escape from the immune response. In the GII.4 genotype, surface-exposed amino acid replacements in the P2 region of the capsid correlate to two distinct phenotypic differences in these viruses. First, variation alters HBGA binding, such that single replacements confer an increased or altered binding capacity22. Second, some of these amino acid replacements correlate to antigenic variability, such that changes probably allowed escape from the predominant protective memory immune response22. In the late 1980s, the GII.4 viruses were probably not the predominant strains, although detailed studies before 1990 are lacking. However, by 1997 the second GII.4 subclade (Grimsby/Lordsdale) evolved with an increased tropism, theoretically allowing this newly emerged epidemic strain to infect a far greater portion of the general population22,23. It is likely that this event was the precipitating factor in establishing herd immunity, which probably became the driving selective force for the emergence of all subsequent novel GII.4 viruses22,23. Therefore, surface variation in the GII.4 genotype seems to be readily accommodated, selectively or simultaneously altering HBGA binding and, perhaps, host susceptibility patterns, thus leading to antigenic variation and escape from herd immunity.
Comparatively, the GI noroviruses seem to have been constrained by a conserved network of amino acids that are probably required for capsid integrity. Although changes did occur, they were only minor, suggesting that the capsid structure required for GI virus function can tolerate only limited variation. Alternatively, GI strains may preferentially target young individuals or only induce short-term immune responses, ameliorating the need for adaptive change32,50.
Different GI VLPs induce considerable cross reactivity to antibodies raised against GI.1-NV50,51,52. These findings suggest that the minimal set of amino acids that is required for structural integrity may encode one or more key immunogenic epitopes, which may therefore generate at least a partial cross receptor blockade against several different GI noroviruses51. Thus, the evolution of novel strains may have been constrained by structural limitations that restricted the number of possible escape mutants. By contrast, the GII genogroup does not seem to encode a surface-exposed conserved domain and has benefited from insertions and deletions in the P2 region that drastically alter sequence composition and provide the structural space for the innovation of novel escape variants (Fig. 4). Interestingly, the GII.3 genotype is sufficiently similar to the GII.4 genotype that GII.3 virus-specific antibodies cross-react with GII.4 viruses, whereas GII.4-specific antibodies bind to GII.3 VLPs but do not block their interaction with the HBGA53. In addition, the GII.3 strain Toronto has been shown to bind to the H3 and A antigens, two HBGAs that are also bound by many of the GII.4 viruses. These results suggest that the GII.3 viruses may have contributed to the generation of herd immunity against the GII.4 noroviruses, and a GII.3 outbreak in the late 1990s may have helped to establish the widespread herd immunity that became the selective force which drove the emergence of the GII.4 viruses as the predominant epidemic norovirus strain53. Additional human challenge and re-challenge studies using characterized viral strains, known host HBGA expression and norovirus immunological assays that are capable of discriminating between closely related strains will be necessary to unravel the complex interplay between the different circulating norovirus strains and the host immune response. The availability of representative capsid proteins from members of each genogroup and genotype at different points of evolution will be essential for unravelling the complex evolutionary epidemiology of norovirus family members.
Immune response
Little is known about the immune response that is elicited following norovirus infection, because the reagents required to study these important questions are largely unavailable. Most information regarding immunity has been gathered from norovirus outbreaks and human challenge studies, which have provided the only available samples with which to study norovirus immunity in humans. Susceptibility studies have revealed that certain individuals are genetically resistant to infection with specific norovirus strains12; however, previous exposure history and the immune response are likely to have a central role in determining infection outcomes following challenge with the virus. Early human challenge and outbreak studies revealed that individuals with high serum or faecal antibody titres to GI.1-NV before challenge were more likely to become infected with the virus than individuals with low pre-existing antibody titres32,54,55,56. Most but not all individuals were resistant to subsequent infection with the same virus 6 months later; but less than half maintained high antibody titres 6 months after a secondary challenge32. In long-term immunity studies, individuals infected with GI.1-NV were all symptomatically reinfected 27–42 months later31. However, some individuals without common pre-exposure factors who were genetically susceptible to GI.1-NV infection never became infected12. These findings are contradictory, in that short-term immunity has been observed and described, whereas persisting long-term immunity has been more difficult to establish.
Studies of seroprevalence to GI and GII strains have shown that at least 50% of children under 5 years of age are seropositive for norovirus exposure; this increases to 60–90% by 10 years of age and reaches 100% by adulthood, demonstrating that nearly all adults have been exposed to one or more noroviruses57,58,59,60,61,62.
Antigenicity and immunity
Population dynamics play an important part in the emergence of novel pathogens, as a population that is largely resistant to one variant, because of herd immunity, provides an environment that can select for an escape from that immunity. In fact, using the influenza virus as a prototype, a model has been generated that suggests that a large epidemic the previous year probably generates enough genetic variation to give rise to a strain that almost completely escapes host immunity63,64. It has been shown that a viral population evolves more rapidly in the presence of a highly immune host population65. This is particularly true for RNA viruses, which generally introduce one mutation per genome per round of viral replication. Indeed, because of their high mutation rate, RNA viruses often exist as a quasispecies in the infected host66,67. In this way, herd immunity is the environment that selects for variation, which may already exist to a lesser extent in the quasispecies.
In the genus Norovirus, confounding factors have contributed to a more complicated evolutionary path. First, different genogroups, genotypes and even viruses in a genotype bind to different HBGAs that are polymorphic in the general population. This indicates that a virus that has been neutralized by herd immunity in one polymorphic population could exploit another susceptible population by differential HBGA ligand recognition. In addition, many noroviruses bind to more than one type of HBGA, suggesting that different populations may be susceptible to one strain. Second, in many cases variation between and within genotypes and genogroups alters antigenicity, which may allow escape from the predominant memory immune response, whereas in other cases cross reactivity may allow partial neutralization or an immune response that is skewed toward a previous exposure (Fig.5).

a | Noroviruses of the GI genogroup seem to be limited in the amount of variation that occurs in the P2 subdomain of the capsid, which reduces the ability of the virus to generate antibody escape mutants. Therefore, viruses in the GI genogroup probably persist by evolving differential binding capacity or through original antigenic sin (OAS) of the host. b | By contrast, the GII genogroup contains more sequence information in the P2 subdomain, which can tolerate more substantial changes, probably allowing evolution to rearrange the surface and alter both binding capacity and antigenic properties. Therefore, as has been shown for GII.4, GII noroviruses are likely to persist by two mechanisms: receptor switching and antigenic drift.
Experiments conducted using a time-ordered panel of GII.4 VLPs showed that this genotype uses both antibody escape and receptor switching to persist in human populations. Sequence analysis identified six unique subclades in the GII.4 genotype, and representative sequences from each were selected and used to generate a panel of time-ordered GII.4 VLPs that spanned from 1987 to 2006. Using a synthetic CHO-binding assay, it was determined that specific amino acid changes proximal to the HBGA interaction site that had occurred over the 20 year interval altered CHO-binding patterns in a subclade-specific manner22,53. In addition, serum from a human outbreak in 1988 and mouse antisera were used to conduct a surrogate neutralization assay using ELISA and in vitro CHO blockade experiments, which found that subclade-specific amino acid replacements altered the serological and blockade responses in a manner that is consistent with a model of antigenic drift22. These results show that the GII.4 viruses adapted to escape herd immunity, to infect previously resistant portions of the polymorphic population or to do both22. Periods of stasis that occurred during the epochal evolution of the GII.4 genotype from approximately 1987 to 1997 and from 1997 to 2002 probably occurred owing to a lack of herd immunity in substantial portions of the polymorphic population; therefore, the selective force necessary to drive the evolution of new epidemic strains was not fully present until approximately 2002. Since 2002, new epidemic strains of GII.4 viruses occur after major outbreaks every 1–2 years, similar to influenza virus emergence, indicating that a vaccine against the GII.4 and perhaps other select norovirus genotypes would require frequent reformulation.
In comparison, the GI genogroup seems to be limited structurally by a surface-exposed conserved domain, which may provide a common antibody epitope in the genogroup. Recent work has demonstrated that the GI noroviruses can bind many different HBGAs, despite the conserved HBGA-binding site (Fig. 5; Table 1). In addition, individuals infected with a GI norovirus generally mount robust B cell and T cell responses against the homologous strain, although some individuals preferentially mount immune responses against heterologous GI strains50. These results corroborate previous findings that have found a high degree of antibody cross reactivity between GI strains68. Cross-reactive B cell and T cell epitopes could allow complex patterns of cross protection among GI strains and promote infectivity by original antigenic sin (OAS), which has been described in mice69,70 and in humans71 at the antibody and T cell levels. Consequently, pre-exposure histories may be a confounding factor in long-term protection against GI strains. Detailed mapping of antigenic epitopes recognizing antibodies or T cells is a crucial need for future norovirus research.
The OAS response generates antibodies and T cells that are specific to the previous pathogen and have reduced affinity toward the novel pathogen72. Although it is currently speculative, an OAS response could account for the results observed in the analysis of VLPs from the GI.1–GI.4 genotypes, in which GI.1-NV-specific antisera cross-reacted with GI.1.2001, GI.2, GI.3 and GI.4 VLPs. This showed that some individuals have a preferential blockade response to other VLPs besides the infecting strain. However, OAS has not been previously shown for noroviruses and is controversial in other viral infections. Despite reports in the literature suggesting OAS as a mechanism in HIV and influenza virus infections72,73, a recent report investigating the rapid cloning of high-affinity human monoclonal antibodies generated by influenza virus vaccination found that nearly all of the vaccine-specific monoclonal antibodies had highest affinity for the vaccine strain74. This observation suggests that OAS does not occur in normal, healthy adults receiving the influenza virus vaccination and argues against OAS as a mechanism that occurs during re-exposure to a viral infection. Alternatively, it could be that OAS responses are complex and only occur in selected individuals who have the appropriate pre-exposure histories; more detailed studies with more individuals are essential to definitively address this question. A similar study using norovirus VLP vaccination of human populations would also provide direct insight into this issue and is warranted.
Challenges to vaccine development
The multiple mechanisms that are used by noroviruses to persist in human populations, along with the complex antigenic diversity and evolution of these viruses, provide substantial challenges to vaccine development. Principle barriers include: the difficulty of finding a vaccine design that provides maximal coverage of the strains that are commonly associated with human disease; the lack of clearly defined measures of protective immunity; the lack of robust animal models for human disease; and the lack of understanding of short-term versus long-term immunity. Studies using mouse and pig models of norovirus pathogenesis have shown that protective immune responses probably target the capsid protein and neutralize infectivity75,76,77,78,79, which suggests that vaccines composed of capsid protein variants would be candidates for human vaccinations. In particular, the pig model is relevant to dissect the effect of antigenic variation on protective immunity against the GII.4 viruses. However, it is still unknown if the protection generated in these models is long term, and further studies are necessary to evaluate the potential impact of pre-exposure history on vaccine efficacy. Extreme antigenic variation exists between various GI and GII strains, and evidence for robust cross protection is lacking, indicating that multivalent VLP vaccine formulations will be essential for broad coverage against the predominant circulating strains. Moreover, until recently many of the different genotypes were treated as a single phenotypic clade, but evolution among the GII.4 viruses means that each subclade must be considered individually. Therefore, it will be essential to define the evolutionary patterns of each genotype in order to identify appropriate contemporary isolates for vaccine design. Although vaccine studies should focus on the predominant GII.4 genotype, control of a single genotype through vaccination would probably leave an evolutionary niche that would be exploited by another GII genotype, perhaps a GII.2 or a GII.3 variant.
Protective cross immunity among the GI genogroup strains may allow for robust vaccines that protect across the entire genogroup; however, empirical support for effective cross-protective immunity is lacking. OAS may further complicate vaccine formulations, as individual patient pre-exposure histories will be nearly impossible to discern and unravel. Furthermore, pre-existing antibodies do not always protect against subsequent infection, and individuals with higher pre-challenge antibody titres have been shown to be more susceptible to infection32,54,55,56. As has been reported for infections with dengue virus and other viruses71,72, the presence of pre-existing antibodies or memory T cells against related but distinct virus strains can decrease the efficacy of the immune response to subsequent infecting strains as well as enhance disease severity. Although a percentage of the human population is genetically resistant to infection with the GI.1-NV strain, which may explain why certain individuals have lower pre-existing antibody titres and are less likely to be infected12, an individual's susceptibility alleles are not capable of controlling infection to all norovirus strains35.
In addition, evolution of the viral capsid protein influences the outcome of infection22. Single amino acid changes in or peripheral to receptor-binding domains in the viral capsid protein enhance, reduce or alter receptor-binding specificities. Because individuals are polymorphous for alleles controlling HBGA expression, altering the viral binding specificities probably allows infection of previously resistant individuals, thereby enabling the virus to escape population-specific herd immunity. These observations suggest that multivalent vaccines composed of several GII.4 capsid antigens would be necessary to control epidemic outbreaks caused by GII.4 noroviruses.
Multivalent regimens containing vaccine components from several predominant circulating strains will possibly protect against subsequent homologous infection, as multivalent VLP vaccines have been shown to protect against homologous infection in the mouse and the pig52,76,77. In addition, it has been shown that increasing the number of strains in multivalent vaccines generates strong and equivalent immune responses to all components of the vaccine without diminishing individual responses compared with the response to a monovalent vaccination51,52.
Furthermore, multivalent vaccines in the mouse have been shown to enhance genogroup-specific, cross-reactive receptor blocking-antibody and T cell responses, and this suggests that multivalent vaccines containing components of both GI and GII strains would probably confer substantial protection against norovirus infection. Promising studies with multivalent Venezuelan equine encephalitis virus replicon particle vaccines against MNV administered to mice showed that immunity remains intact 6 months post-vaccination79, suggesting that long-term immunity can be generated by vaccination. Elegant studies with MNV1 also suggest that neutralizing-antibody and CD4+ T cell or CD8+ T cell responses are essential for protection from reinfection52,79.
In summary, the design of efficacious multivalent norovirus vaccines is essential for reducing the large-scale outbreaks that commonly occur in settings in which close human contact is unavoidable. In addition, understanding the complexity of norovirus evolution and subsequent escape from the immune system has redefined how virologists think about norovirus vaccinology and will continue to direct the field towards a better understanding of how noroviruses survive and persist and how vaccines can be rationally designed to control this formidable viral pathogen.
References
Atmar, R. L. & Estes, M. K. The epidemiologic and clinical importance of norovirus infection. Gastroenterol. Clin. North Am. 35, 275–290 (2006).
CDC. Norovirus activity — United States, 2002. Morb. Mortal. Wkly Rep. 52, 41–45 (2003).
Ike, A. C. et al. Molecular epidemiology of norovirus in outbreaks of gastroenteritis in southwest Germany from 2001 to 2004. J. Clin. Microbiol. 44, 1262–1267 (2006).
Jiang, X. et al. Outbreaks of gastroenteritis in elderly nursing homes and retirement facilities associated with human caliciviruses. J. Med. Virol. 50, 335–341 (1996).
Mead, P. S. et al. Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625 (1999).
Kapikian, A. Z. et al. Visualization by immune electron microscopy of a 27-nm particle associated with acute infectious nonbacterial gastroenteritis. J. Virol. 10, 1075–1081 (1972). This article describes the discovery of Norwalk virus, which was the first norovirus to be recognized.
Kapikian, A. Z. The discovery of the 27-nm Norwalk virus: an historic perspective. J. Infect. Dis. 181, S295–S302 (2000).
Jiang, X., Graham, D., Wang, K. & Estes, M. Norwalk virus genome cloning and characterization. Science 250, 1580–1583 (1990). This paper details the cloning of the Norwalk virus genome for the first time.
Jiang, X., Wang, M., Graham, D. & Estes, M. Expression, self-assembly and antigenicity of the Norwalk virus capsid protein. J. Virol. 66, 6527–6532 (1992). This work shows that recombinant Norwalk virus capsid proteins can self-assemble into virus-like particles using the baculovirus expression system.
Green, K. Y., Lew, J. F., Jiang, X., Kapikian, A. Z. & Estes, M. K. Comparison of the reactivities of baculovirus-expressed recombinant Norwalk virus capsid antigen with those of the native Norwalk virus antigen in serologic assays and some epidemiologic observations. J. Clin. Microbiol. 31, 2185–2191 (1993).
Baric, R. S. et al. Expression and self-assembly of norwalk virus capsid protein from Venezuelan equine encephalitis virus replicons. J. Virol. 76, 3023–3030 (2002). The first formation of Norwalk virus-like particles using the Venezualian equine encephalitis vector expression system.
Lindesmith, L. et al. Human susceptibility and resistance to Norwalk virus infection. Natur Med. 9, 548–553 (2003). This article describes the susceptibility of secretor-positive humans to Norwalk virus, suggesting the presence of a human susceptibility allele.
Wyatt, R. G. et al. Comparison of three agents of acute infectious nonbacterial gastroenteritis by cross-challenge in volunteers. J. Infect. Dis. 129, 709–714 (1974).
Fankhauser, R. L. et al. Epidemiologic and molecular trends of “Norwalk-like viruses” associated with outbreaks of gastroenteritis in the United States. J. Infect. Dis. 186, 1–7 (2002).
Green, S. M., Lambden, P. R., Caul, E. O., Ashley, C. R. & Clarke, I. N. Capsid diversity in small round-structured viruses: molecular characterization of an antigenically distinct human enteric calicivirus. Viru Res. 37, 271–283 (1995).
Karst, S. M. et al. STAT1-dependent innate immunity to a Norwalk-like virus. Science. 299, 1575–1578 (2003).
Oliver, S. L. et al. Molecular characterization of bovine enteric caliciviruses: a distinct third genogroup of noroviruses (Norwalk-like viruses) unlikely to be of risk to humans. J. Virol. 77, 2789–2798 (2003).
Vinje, J. & Koopmans, M. P. Simultaneous detection and genotyping of “Norwalk-like viruses” by oligonucleotide array in a reverse line blot hybridization format. J. Clin. Microbiol. 38, 2595–2601 (2000).
Zheng, D. P. et al. Norovirus classification and proposed strain nomenclature. Virology 346, 312–323 (2006).
Katayama, K. et al. Phylogenetic analysis of the complete genome of 18 Norwalk-like viruses. Virology 299, 225–239 (2002).
Pletneva, M. A., Sosnovtsev, S. V. & Green, K. Y. The genome of hawaii virus and its relationship with other members of the caliciviridae. Virus Genes 23, 5–16 (2001).
Lindesmith, L. C. et al. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5, e31 (2008). This paper discusses the binding and antigenic differences between a panel of GII.4 noroviruses that evolved over 20 years, suggesting that herd immunity may select for novel variants over time.
Donaldson, E. F., Lindesmith, L. C., Lobue, A. D. & Baric, R. S. Norovirus pathogenesis: mechanisms of persistence and immune evasion in human populations. Immunol. Rev. 225, 190–211 (2008).
Siebenga, J. J. et al. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81, 9932–9941 (2007).
Allen, D. J., Gray, J. J., Gallimore, C. I., Xerry, J. & Iturriza-Gomara, M. Analysis of amino acid variation in the P2 domain of the GII-4 norovirus VP1 protein reveals putative variant-specific epitopes. PLoS ONE 3, e1485 (2008).
Iritani, N. et al. Genetic analysis of the capsid gene of genotype GII.2 noroviruses. J. Virol. 82, 7336–7345 (2008).
Carlsson, B. et al. Quasispecies dynamics and molecular evolution of human norovirus capsid P region during chronic infection. J. Gen. Virol. 90, 432–441 (2009).
Hennessy, E. P., Green, A. D., Connor, M. P., Darby, R. & MacDonald, P. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 188, 176–177 (2003).
Hutson, A. M., Atmar, R. L., Graham, D. Y. & Estes, M. K. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 185, 1335–1337 (2002).
Rockx, B. H., Vennema, H., Hoebe, C. J., Duizer, E. & Koopmans, M. P. Association of histo-blood group antigens and susceptibility to norovirus infections. J. Infect. Dis. 191, 749–754 (2005).
Parrino, T. A., Schreiber, D. S., Trier, J. S., Kapikian, A. Z. & Blacklow, N. R. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 297, 86–89 (1977).
Johnson, P. C., Mathewson, J. J., DuPont, H. L. & Greenberg, H. B. Multiple-challenge study of host susceptibility to Norwalk gastroenteritis in US adults. J. Infect. Dis. 161, 18–21 (1990).
Marionneau, S. et al. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122, 1967–1977 (2002).
Thorven, M. et al. A homozygous nonsense mutation (428G→A) in the human secretor (FUT2) gene provides resistance to symptomatic norovirus (GGII) infections. J. Virol. 79, 15351–15355 (2005).
Lindesmith, L. et al. Cellular and humoral immunity following Snow Mountain virus challenge. J. Virol. 79, 2900–2909 (2005).
Larsson, M. M. et al. Antibody prevalence and titer to norovirus (genogroup II) correlate with secretor (FUT2) but not with ABO phenotype or Lewis (FUT3) genotype. J. Infect. Dis. 194, 1422–1427 (2006).
Huang, P. et al. Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major binding groups among multiple binding patterns. J. Virol. 79, 6714–6722 (2005).
Carlsson, B. et al. The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 norovirus infection. PLoS ONE 4, e5593 (2009).
Shirato, H. et al. Noroviruses distinguish between type 1 and type 2 histo-blood group antigens for binding. J. Virol. 82, 10756–10767 (2008).
Rydell, G. E. et al. Human noroviruses recognize sialyl Lewis x neoglycoproteins. Glycobiology 19, 309–320 (2009).
Prasad, B. V., Rothnagel, R., Jiang, X. & Estes, M. K. Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J. Virol. 68, 5117–5125 (1994).
Prasad, B. V. et al. X-ray crystallographic structure of the Norwalk virus capsid. Science 286, 287–290 (1999). This work describes the three-dimensional structure of the Norwalk VLP.
Bertolotti-Ciarlet, A., White, L. J., Chen, R., Prasad, B. V. & Estes, M. K. Structural requirements for the assembly of Norwalk virus-like particles. J. Virol. 76, 4044–4055 (2002).
Bu, W. et al. Structural basis for the receptor binding specificity of the Norwalk virus. J. Virol. 82, 5340–5347 (2008).
Choi, J. M., Hutson, A. M., Estes, M. K. & Prasad, B. V. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl Acad. Sci. USA 105, 9175–9180 (2008). This study and reference 44 demonstrate the three-dimensional structure of the Norwalk virus's protruding domain in complex with different HBGAs.
Prasad, B. V., Hardy, M. E. & Estes, M. K. Structural studies of recombinant Norwalk capsids. J. Infect. Dis. 181, S317–S321 (2000).
Tan, M. et al. Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: evidence for a binding pocket. J. Virol. 77, 12562–12571 (2003).
Cao, S. et al. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 81, 5949–5957 (2007). This study elucidates the crystal structure of the protruding domain of a GII.4 virus in complex with HBGAs.
Nilsson, M. et al. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 77, 13117–13124 (2003).
Lindesmith, L. C. et al. Heterotypic humoral and cellular immune responses following Norwalk virus infection. J. Virol. 9 Dec 2009 (doi:10.1128/JVI.02179-09).
LoBue, A. D. et al. Multivalent norovirus vaccines induce strong mucosal and systemic blocking antibodies against multiple strains. Vaccine 24, 5220–5234 (2006).
LoBue, A. D., Thompson, J. M., Lindesmith, L., Johnston, R. E. & Baric, R. S. Alphavirus-adjuvanted norovirus-like particle vaccines: heterologous, humoral, and mucosal immune responses protect against murine norovirus challenge. J. Virol. 83, 3212–3227 (2009).
Cannon, J. L. et al. Herd immunity to GII.4 noroviruses is supported by outbreak patient sera. J. Virol. 83, 5363–5374 (2009).
Gray, J. J. et al. Detection of immunoglobulin M (IgM), IgA, and IgG Norwalk virus-specific antibodies by indirect enzyme-linked immunosorbent assay with baculovirus-expressed Norwalk virus capsid antigen in adult volunteers challenged with Norwalk virus. J. Clin. Microbiol. 32, 3059–3063 (1994).
Okhuysen, P. C., Jiang, X., Ye, L., Johnson, P. C. & Estes, M. K. Viral shedding and fecal IgA response after Norwalk virus infection. J. Infect. Dis. 171, 566–569 (1995).
Baron, R. C., Greenberg, H. B., Cukor, G. & Blacklow, N. R. Serological responses among teenagers after natural exposure to Norwalk virus. J. Infect. Dis. 150, 531–534 (1984).
Cubitt, W. D., Green, K. Y. & Payment, P. Prevalence of antibodies to the Hawaii strain of human calicivirus as measured by a recombinant protein based immunoassay. J. Med. Virol. 54, 135–139 (1998).
Hinkula, J., Ball, J. M., Lofgren, S., Estes, M. K. & Svensson, L. Antibody prevalence and immunoglobulin IgG subclass pattern to Norwalk virus in Sweden. J. Med. Virol. 47, 52–57 (1995).
Honma, S. et al. Epidemiological study of prevalence of genogroup II human calicivirus (Mexico virus) infections in Japan and Southeast Asia as determined by enzyme-linked immunosorbent assays. J. Clin. Microbiol. 36, 2481–2484 (1998).
Jing, Y., Qian, Y., Huo, Y., Wang, L. P. & Jiang, X. Seroprevalence against Norwalk-like human caliciviruses in Beijing, China. J. Med. Virol. 60, 97–101 (2000).
Nakata, S. et al. Prevalence of human calicivirus infections in Kenya as determined by enzyme immunoassays for three genogroups of the virus. J. Clin. Microbiol. 36, 3160–3163 (1998).
Parker, S. P., Cubitt, W. D. & Jiang, X. Enzyme immunoassay using baculovirus-expressed human calicivirus (Mexico) for the measurement of IgG responses and determining its seroprevalence in London, UK. J. Med. Virol. 46, 194–200 (1995).
Boni, M. F. Vaccination and antigenic drift in influenza. Vaccine 26 (Suppl. 3), C8–C14 (2008).
Boni, M. F., Gog, J. R., Andreasen, V. & Christiansen, F. B. Influenza drift and epidemic size: the race between generating and escaping immunity. Theor. Popul. Biol. 65, 179–191 (2004).
Boni, M. F., Gog, J. R., Andreasen, V. & Feldman, M. W. Epidemic dynamics and antigenic evolution in a single season of influenza A. Proc. Biol. Sci. 273, 1307–1316 (2006).
Domingo, E. RNA virus evolution and the control of viral disease. Prog. Drug Res 33, 93–133 (1989).
Eigen, M. The origin of genetic information: viruses as models. Gene 135, 37–47 (1993).
Noel, J. S. et al. Correlation of patient immune responses with genetically characterized small round-structured viruses involved in outbreaks of nonbacterial acute gastroenteritis in the United States, 1990 to 1995. J. Med. Virol. 53, 372–383 (1997).
Beaumier, C. M., Mathew, A., Bashyam, H. S. & Rothman, A. L. Cross-reactive memory CD8+ T cells alter the immune response to heterologous secondary dengue virus infections in mice in a sequence-specific manner. J. Infect. Dis. 197, 608–617 (2008).
Larke, N. et al. Combined single-clade candidate HIV-1 vaccines induce T cell responses limited by multiple forms of in vivo immune interference. Eur. J. Immunol. 37, 566–577 (2007).
Mongkolsapaya, J. et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nature Med. 9, 921–927 (2003).
Muller, S. Avoiding deceptive imprinting of the immune response to HIV-1 infection in vaccine development. Int. Rev. Immunol. 23, 423–436 (2004).
Tobin, G. J. et al. Deceptive imprinting and immune refocusing in vaccine design. Vaccine 26, 6189–6199 (2008).
Wrammert, J. et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008).
Souza, M., Azevedo, M. S., Jung, K., Cheetham, S. & Saif, L. J. Pathogenesis and immune responses in gnotobiotic calves after infection with the genogroup II.4-HS66 strain of human norovirus. J. Virol. 82, 1777–1786 (2008).
Souza, M., Costantini, V., Azevedo, M. S. & Saif, L. J. A human norovirus-like particle vaccine adjuvanted with ISCOM or mLT induces cytokine and antibody responses and protection to the homologous GII.4 human norovirus in a gnotobiotic pig disease model. Vaccine 25, 8448–8459 (2007).
Souza, M., Cheetham, S. M., Azevedo, M. S., Costantini, V. & Saif, L. J. Cytokine and antibody responses in gnotobiotic pigs after infection with human norovirus genogroup II.4 (HS66 strain). J. Virol. 81, 9183–9192 (2007).
Chachu, K. A. et al. Antibody is critical for the clearance of murine norovirus infection. J. Virol. 82, 6610–6617 (2008).
Chachu, K. A., LoBue, A. D., Strong, D. W., Baric, R. S. & Virgin, H. W. Immune mechanisms responsible for vaccination against and clearance of mucosal and lymphatic norovirus infection. PLoS Pathog. 4, e1000236 (2008).
Green, J., Norcott, J. P., Lewis, D., Arnold, C. & Brown, D. W. Norwalk-like viruses: demonstration of genomic diversity by polymerase chain reaction. J. Clin. Microbiol. 31, 3007–3012 (1993).
Caley, I. J. et al. Humoral, mucosal, and cellular immunity in response to a human immunodeficiency virus type 1 immunogen expressed by a Venezuelan equine encephalitis virus vaccine vector. J. Virol. 71, 3031–3038 (1997).
Davis, N. L., Brown, K. W. & Johnston, R. E. A viral vaccine vector that expresses foreign genes in lymph nodes and protects against mucosal challenge. J. Virol. 70, 3781–3787 (1996).
Davis, N. L. et al. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine encephalitis virus replicon particles. J. Virol. 74, 371–378 (2000); erratum 74, 3430 (2000).
Pushko, P. et al. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 239, 389–401 (1997).
Caley, I. J. et al. Venezuelan equine encephalitis virus vectors expressing HIV-1 proteins: vector design strategies for improved vaccine efficacy. Vaccine 17, 3124–3135 (1999).
Kamrud, K. I. et al. Analysis of Venezuelan equine encephalitis replicon particles packaged in different coats. PLoS ONE 3, e2709 (2008).
Davis, N. L. et al. Alphavirus replicon particles as candidate HIV vaccines. IUBMB Life 53, 209–211 (2002).
Rayner, J. O., Dryga, S. A. & Kamrud, K. I. Alphavirus vectors and vaccination. Rev. Med. Virol. 12, 279–296 (2002).
Someya, Y. Genomic organization of Norwalk-like viruses and functions of viral gene products. Nippon Rinsho 60, 1155–1164 (2002) (in Japanese).
Huelsenbeck, J. P., R. F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Marti-Renom, M. A. et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 (2000).
Acknowledgements
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health (grant AID56351).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Caliciviridae
-
A family of small (2740 nm), non-enveloped, icosahedral enteric viruses that infect a broad range of host species. These positive-sense, single-stranded RNA viruses obtained their name from the Latin word calix, meaning chalice, owing to the cup-shaped structures on the surface of intact capsids. Caliciviruses are divided into four genera: Norovirus, Sapovirus, Vesiviruses and Lagoviruses.
- Virus-like particle
-
A non-infectious particle formed by viral structural proteins that recapitulates the virus structure without packaging the viral nucleic acid or proteins that are normally packaged by the virus.
- Protective immunity
-
An acquired immune response sufficient to protect an individual from infection or reinfection by a pathogen. It can be induced by vaccination or by previous exposure to a particular pathogen.
- Quasispecies
-
A cloud of genetically similar viral genotypes, some of which may contain a less fit phenotype that is capable of exploiting a new niche that is created by the neutralization of the predominant viral phenotype.
- Herd immunity
-
Immunity that occurs in a population when enough individuals become immune to a particular pathogen to break the transmission chain of that pathogen, providing pseudo-protection to non-immune individuals.
- Histo-blood group antigen
-
A member of the family of complex glycans that are expressed on the surfaces of red blood cells, gut and respiratory epithelia, and biological secretions in humans. Their expression is regulated by the fucosyltransferase genesFUT1, FUT2 and FUT3, which are polymorphic in human populations. Differential expression of these enzymes determines the moieties and modifications to these ligands that are expressed by different humans.
- Secretor-positive
-
Pertaining to an individual who expresses the FUT1 ABO blood group antigens on the cells that line their mouths and guts and whose saliva contains the same antigens.
- Non-secretor
-
Pertaining to an individual who does not express the FUT1 ABO blood group antigens on the cells that line their mouths and guts and therefore do not secrete them in the saliva.
- Triangulation number
-
A measure that is used to describe the structure of the icosahedral viral capsid and that was first described by Caspar and Klug. The icosahedron itself has 20 equilateral triangular facets, each of which is divided into a number (T) of identical equilateral triangular subdivisions (or protein subunits), such that the capsid has 20T structure subunits.
- Original antigenic sin
-
A host-mediated humoral or cellular immune response in which a memory response against a previously encountered pathogen is preferentially used against a new pathogen variant rather than another primary response being produced.
Rights and permissions
About this article
Cite this article
Donaldson, E., Lindesmith, L., LoBue, A. et al. Viral shape-shifting: norovirus evasion of the human immune system. Nat Rev Microbiol 8, 231–241 (2010). https://doi.org/10.1038/nrmicro2296
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2296
This article is cited by
-
Influence of herd immunity on norovirus: a long-term field study of repeated viral gastroenteritis outbreaks at the same facilities
BMC Infectious Diseases (2023)
-
Molecular epidemiology analysis of symptomatic and asymptomatic norovirus infections in Chinese infants
Virology Journal (2023)
-
Recent trends and developments of PCR-based methods for the detection of food-borne Salmonella bacteria and Norovirus
Journal of Food Science and Technology (2022)
-
Molecular evolution of GII.P17-GII.17 norovirus associated with sporadic acute gastroenteritis cases during 2013–2018 in Zhoushan Islands, China
Virus Genes (2020)
-
Effect of natural ageing and heat treatments on GII.4 norovirus binding to Histo-Blood Group Antigens
Scientific Reports (2019)
