
Key Points
-
There have been recent advances in the methodologies used for isolating and producing antigen-specific monoclonal antibodies that are naturally generated in humans in response to vaccines or infections. This has allowed a rapid and productive rise in the isolation and characterization of fully human monoclonal antibodies.
-
These human monoclonal antibodies are greatly improving our knowledge of the natural human response to pathogens and are instrumental in epitope discovery. They are also being developed as therapeutic agents against many infectious and autoimmune diseases.
-
Three different strategies have been used to identify and isolate B cells expressing immunoglobulins with the desired specificity and functional characteristics at the monoclonal level. The first involves panning phage display libraries that have been constructed from the immunoglobulin variable genes of immunized or infected individuals. In the second approach, memory B cells are immortalized, and then in vitro cultures are screened for antibody specificity. The third method involves single-cell sorting, followed by cloning of the transcribed immunoglobulin genes and their expression as monoclonal antibodies; this strategy may or may not include flow cytometry-based pre-selection.
-
If the intent is to isolate the most effective neutralizing human monoclonal antibodies, then highly targeted high-throughput screening is the most appropriate strategy. This can be achieved through phage display, memory B cell immortalization or flow cytometry-based antigen-specific selection from an immune individual. To fully characterize the spectrum of the B cell repertoire responding to an immune challenge, broader, less selective criteria can be used for cloning human monoclonal antibodies.
-
The most recent and exciting advances in the isolation of human monoclonal antibodies have been in response to HIV and influenza virus infection or vaccination. Clever antibody-screening methods and the careful selection of human donors have allowed for the isolation of rare, broadly neutralizing antibodies to both of these viruses.
-
It is hoped that these recent advances in isolating naturally generated broadly neutralizing antibodies specific for evolving viruses will speed up the development of effective vaccines.
Abstract
The natural human antibody response is a rich source of highly specific, neutralizing and self-tolerant therapeutic reagents. Recent advances have been made in isolating and characterizing monoclonal antibodies that are generated in response to natural infection or vaccination. Studies of the human antibody response have led to the discovery of crucial epitopes that could serve as new targets in vaccine design and in the creation of potentially powerful immunotherapies. With a focus on influenza virus and HIV, herein we summarize the technological tools used to identify and characterize human monoclonal antibodies and describe how these tools might be used to fight infectious diseases.
Similar content being viewed by others

Main
Since the late nineteenth century, naturally produced human serum immunoglobulins have been used as a passive immunotherapy for a wide range of infectious diseases1. However, owing to the polyclonal and variable nature of serum immunoglobulins, these therapies have had mixed efficacies, and they are also hindered by the added risk of blood-borne pathogen transmission, not to mention the expense and practical limits of the large-scale production of human blood products.
The development of hybridoma technology by César Milstein's group in the mid-1970s allowed for the production of mouse monoclonal antibodies with defined specificities and neutralization capacities2. Although these mouse monoclonal antibodies revolutionized biomedical sciences, they proved to be problematic for human therapeutic use, as they had a short serum half-life in humans and they were often immunogenic with low effector function3. With the advent of technologies to produce humanized and chimeric monoclonal antibodies, many of these problems were overcome, leading to the generation of a number of monoclonal antibodies that are currently approved for clinical use4.
In recent years, however, the development of methodologies for producing antigen-specific monoclonal antibodies directly from human B cells has allowed a rapid and productive rise in the isolation and characterization of fully human monoclonal antibodies that are naturally generated in response to autoantigens or pathogens. Because these monoclonal antibodies are produced and tolerized in humans, their safety, efficacy and relevance to human disease are increased compared with antibodies generated in mice or other species. These human monoclonal antibodies are proving invaluable in improving our knowledge of the natural human response to pathogens and for epitope discovery. Human monoclonal antibodies also have a high potential to serve as potent therapeutic tools against a range of infectious and non-infectious diseases. For example, until recently, it was thought that certain single antibodies capable of neutralizing a broad array of evolving viruses, such as influenza virus or HIV strains, were extremely rare and almost impossible to isolate. However, high-throughput technologies, clever screening processes and the careful selection of infected donors have now allowed for the isolation and characterization of several broadly neutralizing monoclonal antibodies. This has led to the discovery of newly appreciated conserved epitopes that could be targeted in antibody-mediated therapy and/or could guide vaccine design.
There is now a growing optimism that future work will lead to the discovery of additional epitopes that can be targeted to fight many infectious diseases. Characterization of the antibodies themselves has also led to new insights into how, at a protein structural level, antibodies can be generated that can access epitopes masked by immune evasion mechanisms. All together, scrutiny of the natural antibody response has led to unparalleled insights into the unique strategies used by the human humoral system to neutralize pathogens.
In this Review, we focus on human monoclonal antibodies that have been derived directly from human B cells that were generated in vivo in response to specific infections, rather than on antibodies derived from synthetic immunoglobulin recombinant libraries or humanized mouse systems. Although these latter technologies have generated valuable monoclonal antibodies that are used therapeutically in cancer, metabolic diseases and autoimmune diseases (as reviewed elsewhere5,6,7), they are beyond the scope of this Review. Here, we summarize the current technologies used to isolate human monoclonal antibodies and describe how these technologies are being applied to understand the natural immune response to influenza virus and HIV. Recent advances in the isolation and characterization of broadly neutralizing human monoclonal antibodies from individuals exposed to these viruses have elegantly illustrated the power and potential of characterizing the natural humoral immune response. The varied approaches used to characterize human monoclonal antibodies specific for influenza virus and HIV provide a cross-section of the technologies being used to study human monoclonal antibodies and of the uses for the knowledge gained. However, these same methods can be used to make therapeutics and improved vaccines for other infectious diseases, such as hepatitis C virus, dengue virus, pneumococcal and staphylococcal bacteria, and any other pathogen that antibodies can control.
Finding the right B cell: a needle in a haystack
Activated B cells differentiate into memory B cells and antibody-secreting cells, which can be further categorized into short-lived plasmablasts and long-lived plasma cells. Both memory and antibody-secreting B cell populations have been used to generate naturally derived antigen-specific monoclonal antibodies.
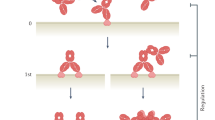
The main obstacle to the generation of such monoclonal antibodies has been in identifying and isolating B cells expressing immunoglobulins with the desired specificity and functional characteristics at the single-cell level. Broadly speaking, three different strategies have been used to do this (Fig. 1). First, and most classically, many antibodies have been isolated through the panning of phage display libraries constructed from the immunoglobulin variable genes of immunized or infected individuals. A second common approach is B cell immortalization followed by the screening of in vitro cultures for antibody specificity. More recently, a third approach has been developed that uses single-cell sorting followed by cloning of the transcribed immunoglobulin genes and their expression as monoclonal antibodies, with or without flow cytometry-based pre-selection.

Three general approaches and variations therein are typically used to isolate monoclonal antibodies from humans. The particular method used depends on various factors but is largely driven by the types of samples obtained. a | Samples with rare specific memory B cells require a higher throughput methodology, such as phage display, to identify the rarest specificities. b | Epstein–Barr virus (EBV)-mediated transformation or other B cell immortalization strategies can be used for most samples in which an immunological history is known. c | In instances in which an effective antigen bait is available that will allow for the detection of B cells with specific receptors using flow cytometry, single-cell expression cloning can be used. Finally, if there is an ongoing or recent immune response to vaccination or infection then the population of activated plasmablasts will contain a high frequency of specific cells that can be used to generate abundant human monoclonal antibodies by expression cloning.
These different selection methods are more or less amenable to different B cell subtypes. Phage display libraries have been successfully generated using sorted memory B cells and total peripheral blood mononuclear cell populations containing all B cell subtypes, including activated and naive B cells. Memory B cells are the most amenable B cell subset to immortalization and in vitro culture, after which they can be screened for antibody specificity, although it has been recently demonstrated that single plasmablasts can be cultured for a long enough time for them to secrete sufficient amounts of antibody to enable screening for multiple antibody specificities without immortalization8,9. Memory B cells have also been successfully differentiated into antibody-secreting cells without immortalization, and the secreted antibodies are then screened for function and specificity10. The detection of specific B cells by flow cytometry requires the expression of immunoglobulins on the cell surface and so has worked well with memory B cells and IgA+ mucosal long-lived plasma cells. However, this selection method has been less successful with IgG+ plasma cells and plasmablasts in peripheral blood, as these cells have low surface immunoglobulin levels11. Because plasmablasts are activated by ongoing immune responses and are therefore enriched for the antibody specificity of interest, this cell type is normally isolated without pre-screening, and antigen-specific human monoclonal antibodies are identified after cloning and expression. Below we provide more detail on these isolation methods, their advantages and disadvantages (Table 1), and the diseases that have been targeted by each of these various approaches (Table 2).
High-throughput phage display technologies. Phage display libraries of single-chain variable antibody fragments (scFvs) or antigen-binding fragments (Fabs) have been used extensively to identify and clone monoclonal antibodies with a multitude of specificities. The construction of the variable gene libraries from B cells isolated from immunized individuals or from individuals post infection can serve as a way to search in a high-throughput manner for antibodies produced by B cells responding to a specific pathogen. This approach has been useful in isolating neutralizing antibodies specific for West Nile virus, rabies virus, severe acute respiratory syndrome (SARS) virus, hepatitis A virus, HIV, hantavirus, Ebola virus, yellow fever virus, hepatitis C virus, measles virus and human and avian influenza virus strains, as well as for other pathogens, using libraries generated from bone marrow, peripheral blood and splenic human B cells from immunized or infected individuals12.
While this has been an excellent method for obtaining neutralizing antibodies to a myriad of diseases, the resulting library of antibody fragments is not necessarily a faithful representation of the physiological antibody gene pairs, as the antibody fragments are generated from the random pairing of immunoglobulin heavy and light chain variable regions that are cloned separately. Thus, with phage display it is not certain that a given heavy and light chain pair went through selection and is self-tolerant. However, this random pairing also creates a greater diversity of antibodies, and the shuffling of the heavy and light chain repertoires has been used to generate higher affinity antibodies13. Phage display also makes it difficult to draw direct conclusions as to the relative proportion of particular antibody specificities and to gain an understanding of the complete repertoire of antigen-specific B cells. Furthermore, antibody expression in the phage display system may not be the same as in mammalian cells owing to differences in protein folding and post-translational modifications, causing a bias against the isolation of some antibodies. To mitigate this problem to some extent, systems have also been developed in which recombinant antibody fragments are expressed on the cell surface of yeast or mammalian cells, and antigen-specific cells are then selected using flow-based methods7.
B cell immortalization. Human B cells proved to be more difficult to immortalize using hybridoma technology than mouse B cells. However, an alternative approach first published in 1977 (Ref. 14) was to immortalize human memory B cells using Epstein–Barr virus (EBV)-mediated transformation. Although some human monoclonal antibodies were produced in the 1980s and 1990s using this method15,16,17,18,19, the inefficiency of the method made it difficult for use in generating large numbers of antigen-specific B cells. Subsequently, however, it became clear that transformation efficiencies could be substantially improved by first activating the B cells using the Toll-like receptor 9 (TLR9) agonist CpG DNA or other polyclonal stimulants before and during EBV infection20. This enhanced EBV transformation protocol or variations of the method have now been widely used for the production of monoclonal antibodies against various pathogenic viruses, including influenza virus21,22,23,24, HIV10,25,26,27, SARS virus20, human cytomegalovirus (HCMV)28, dengue virus29,30,31,32,33,34, chikungunya virus (CHIKV)35 and respiratory syncytial virus (RSV)36.
B cell immortalization typically involves culturing total peripheral blood lymphocytes or sorted IgG+ memory B cells from fresh or frozen samples in the presence of EBV together with a TLR9 ligand and/or allogeneic irradiated mononuclear cells to provide further co-stimulatory signals. Under these conditions, B cells proliferate and secrete antibodies. A week or two later, supernatants from the cultured cells are tested for antigen binding and/or viral neutralization. The B cells producing the antibodies of interest are cloned by limiting dilution and further screened for the desired reactivity at the single-cell level before individual immunoglobulin heavy and light chain pairs are cloned and sequenced20. EBV-transformed B cell clones have also been fused with myeloma cells to generate hybridomas, which facilitates the stable production of high levels of antibodies31,37. Kwakkenbos et al. took a different approach to immortalizing B cells, by transducing memory B cells obtained from peripheral blood with a retrovirus encoding the anti-apoptotic factors B cell lymphoma 6 (BCL-6) and BCL-XL38. In the presence of interleukin-21 (IL-21) and CD40 ligand, these transduced cells differentiated into long-lived antibody-secreting cells that still expressed a B cell receptor on the cell surface, thus allowing selection for antigen specificity by flow cytometry and the screening of secreted antibodies. These cells also maintained activation-induced cytidine deaminase (AID)-mediated mutational activity. Although in vitro mutations may be a source of antibody clone instability, they may also lead to the production of subclones with higher affinities38.
The immortalization of memory B cells and high-throughput screening for antigen-specific B cells has allowed the isolation of rare B cells from the memory pool months or even years after antigenic exposure. For example, EBV-mediated immortalization of memory B cell populations that were isolated from older individuals and had been generated by the immune system decades earlier has allowed the isolation of antibodies specific for influenza virus strains that are no longer in circulation, including the pandemic 1918 H1N1 Spanish flu strain24 and the pandemic H2N2 strain23 that has not been in circulation since 1968. The drawback of the approach is that it involves screening large numbers of B cells to obtain monoclonal antibodies of the desired specificity. Typically, tens of thousands of memory B cells are cultured and screened to obtain less than ten specific B cells.
Single-cell expression cloning. An important advance in human monoclonal antibody technology was the use of single-cell reverse transcription PCR (RT-PCR) to isolate the cognate immunoglobulin heavy and light chain variable genes from single B cells sorted by flow cytometry. These genes could then be cloned and expressed in eukaryotic cell lines. This approach allows human monoclonal antibodies to be obtained even from rare, highly discrete B cell subpopulations, provided that the cells can be identified by flow cytometry. Wardemann et al. were the first to apply this approach to understanding the tolerogenic selection of developing human B cell subpopulations39. This methodology has subsequently been applied to understanding human B cell selection in multiple situations in healthy and immunocompromised individuals40,41,42,43,44,45 and is a central component of the antigen baiting and plasmablast approaches described below.
As an alternative to screening antibodies secreted from antigen-specific B cells in vitro, antigen baiting involves the use of fluorescently labelled antigens to first sort antigen-specific B cells by flow cytometry. First used in combination with B cell immortalization46,47, this approach has now been combined to great effect with single-cell expression cloning48,49,50,51. The method involves cloning the variable regions that have been amplified from single antigen-specific cells into mammalian expression vectors containing constant region genes; these vectors are then used to transfect HEK293 cells for antibody production and purification45,48. The monoclonal antibodies produced are then tested for antigen specificity and neutralizing capacity.
Antigen baiting has been quite successful in obtaining HIV-specific human monoclonal antibodies from the IgG+ memory B cell pools of infected donors using HIV-1 envelope spike proteins as probes52,53,54,55,56. Furthermore, because long-lived IgA+ plasma cells in the mucosal tissue lining the intestine express immunoglobulins on their cell surface11,50,51, antigen baiting has allowed antigen-specific plasma cells to be isolated from intestinal biopsies to generate rotavirus-specific human monoclonal antibodies from healthy individuals50 and transglutaminase 2-specific human monoclonal antibodies from patients with coeliac disease51. The detection of antigen-specific monoclonal antibodies using flow cytometry is limited to B cell types expressing immunoglobulins on the cell surface and by the availability of highly specific and stable antigens to act as probes. However, it allows for very efficient and highly selective isolation of B cells, with 80–90% of the cells sorted by flow cytometry producing immunoglobulins with the desired specificity51,52. Although this method primarily screens for binding specificities and not functional characteristics, as exemplified for studies of HIV-specific human monoclonal antibodies (see below), B cells expressing neutralizing antibodies can be preferentially targeted by cleverly designing the antigen probe.
Owing to the resistance of short-lived plasmablasts to EBV-mediated immortalization and their low surface immunoglobulin expression levels, screening for the immunoglobulin specificity of this B cell type at the monoclonal level has been difficult. Although the efficacy of vaccination has traditionally been analysed by testing the polyclonal serum immunoglobulin response, the cloning and expression of the immunoglobulins from plasmablasts at the monoclonal level provides a new level of resolution to the ongoing immune response and can be a valuable source of therapeutically relevant human monoclonal antibodies33,45,57,58,59. The observation that there is a transient, but often large, population of antigen-specific plasmablasts in peripheral blood 5–8 days after vaccination60,61,62 created the opportunity to sort and clone the immunoglobulin genes of these plasmablasts, from which the majority of immunoglobulins will be antigen specific, and to use these genes for human monoclonal antibody production45,63. This approach is particularly useful for examining the ongoing immune response to vaccines, because the time of antigen exposure is well defined. For example, this method has been successful for analysing the immune response to influenza, tetanus and anthrax vaccination45,57,63,64,65,66,67. The approach works best with freshly isolated cells; although frozen samples have been used68, the survival of plasmablasts is negatively affected by the freezing and thawing process. Random cloning of monoclonal antibodies from activated plasmablasts without pre-screening for specificity can also be inefficient if the starting population contains only a few cells that bind to the antigen of interest. Pre-screening does, however, inherently bias the obtainable information about the antibody response to a given pathogen or autoantigen. Thus, isolation and careful characterization of all plasmablasts can provide unexpected and novel insights into the immune response.
Finally, various high-throughput technologies are being developed to generate a more comprehensive picture of the human B cell response69. All the methods described to this point result in the isolation and characterization of only a small subset of antigen-specific B cells. One promising approach is the use of next-generation sequencing to exhaustively sequence the entire B cell repertoire. By itself, next-generation sequencing can only be used to look at general characteristics of immunoglobulin diversity and usage, as cognate heavy and light chain pairing information is not available. However, the immunoglobulin variable gene sequences derived from next-generation sequencing of a given individual can be mined for B cell sequences that are clonally related to those isolated and characterized using the monoclonal antibody methods described above70. This can provide information on the clonal diversity and affinity maturation of antigen-specific B cells. By varying the time point and the B cell population sequenced, the longevity of a particular B cell specificity of interest and the B cell subsets containing that specificity can also be determined70. Pairing mass spectrometry data obtained from digested serum antibody fragments with next-generation sequencing information from the antibody-secreting cells has also been used recently to resolve at a monoclonal level the polyclonal serum response, thus characterizing the antibody response at both the cellular and the serological level in the same individual53,71. Microarray technologies are also being developed for functional profiling of antibody-secreting cells at the single-cell level and for epitope mapping of serological responses. These new high-throughput technologies have the potential to increase both our efficiency at producing human monoclonal antibodies and the resolution with which these antibodies can be characterized69.
Choosing a method. The choice of method for the isolation of human monoclonal antibodies is determined by several factors. The most important factors include the expertise of the particular investigator and the primary purpose for making human monoclonal antibodies. Furthermore, the state of knowledge that already exists about the antibody response in that particular disease will often dictate the appropriate approach.
If the intent is to isolate for therapeutic purposes human monoclonal antibodies with the highest possible efficacy at neutralizing a given pathogen, then a highly targeted screening of memory B cells for specific binding or a functional characteristic is likely to be the most appropriate approach. This screening can be accomplished through phage display, by testing antibodies secreted in in vitro culture systems, or through flow cytometry-based selection from an immune individual. However, if the purpose is first to fully characterize the spectrum of the B cell repertoire responding to a given antigen or in a particular disease, then broader, less selective criteria can be used for cloning human monoclonal antibodies from both memory and antibody-secreting cells. More-random sampling of the human monoclonal antibody repertoire can provide valuable information on both protective and non-protective immune responses to vaccination or infection. This less-selective approach can also be used to characterize novel neutralizing epitopes or to define autoantibodies that arise in autoimmune disease. As human monoclonal antibodies are isolated and characterized, the information gained from these antibodies can inform subsequent studies and allow fine-tuning of the selection criteria used for isolating new relevant human monoclonal antibodies.
Human antibody response to viral infections
The B cell response to viral infections has been particularly well studied using a combination of the technologies described above and illustrates well the complementary information that the various methods can provide. Here we describe in more detail how the isolation of naturally produced human monoclonal antibodies using different methods has expanded our knowledge of the human immune response to the evolving viral pathogens influenza virus and HIV (Fig. 2).

To maximize the chances of identifying relevant human monoclonal antibodies with both potent and broad reactivity to variant forms of a particular virus or other pathogen, a number of issues must be taken into consideration when the studies are designed. The factors summarized in this figure are detailed within the body text and include the following. a | First, an appropriate cohort must be chosen, such as HIV long-term non-progressors, influenza pandemic survivors, currently or recently infected individuals, vaccinated individuals, and people with a wider breadth or higher potency of serum neutralization than the general population. b | Second, an appropriate methodology must be chosen to generate a library of monoclonal antibodies. c | Third, the relevant resources, reagents and methodologies must be applied to characterize the activity of the antibodies isolated. Examples of such characterizations include: screening for appropriate binding characteristics using enzyme-linked immunosorbent assays (ELISAs) and surface plasmon resonance; determining the breadth and potency of neutralization activity in vitro; testing the in vivo efficacy of protective antibodies in animal models; and mapping epitopes by escape-mutant analysis, target-antigen mutagenesis, competition assays using monoclonal antibodies of known specificity, functional assays and X-ray crystallography. The space-filling diagram denoting the structure of an antibody Fab fragment bound to influenza virus haemagglutinin exemplifies how epitopes are determined using crystallography. d | The biological information gleaned from analysing the highly unique antibody variable gene sequences can be used to further understand the immune responses, as well as to identify additional antibodies that have similar specificities but that differ in fine specificity and efficacy owing to differentially accumulated immunoglobulin somatic mutations. The graph depicts typical nucleotide sequence data.
Influenza virus. The influenza virus is continually evolving and adapting to escape protective immunity, making it difficult to generate a universal vaccine that could provide permanent protection against all influenza virus strains. Worldwide, influenza causes hundreds of thousands of deaths each year owing to local influenza epidemics. The 2009 H1N1 pandemic infected a large portion of the human population, and infection with the avian influenza virus H5N1 resulted in a particularly high mortality rate. These events have led to renewed fear of an influenza pandemic similar to the 1918 Spanish flu that killed 50 to 100 million people. There is now a great deal of interest in finding protective monoclonal antibodies that can act as a passive immunotherapy or that can be used to identify epitopes that can be targeted to provide broad protection across widely divergent influenza strains.
Antibodies that neutralize influenza virus bind to either haemagglutinin or neuraminidase surface glycoproteins, inhibiting viral entry and exit, respectively72. There are 16 different known haemagglutinin subtypes of influenza A virus strains, which can be subdivided into two main phylogenetic groups (group 1 and group 2). The HA1 subunit of haemagglutinin predominantly forms the globular head and contains the sialic acid binding site, and the HA2 subunit primarily forms the stalk and membrane-attachment portion of the molecule (Fig. 3a). With the exception of a broadly neutralizing monoclonal antibody isolated from an immunized mouse73, most neutralizing antibodies that were described before 2008 bound to the globular head HA1 hypervariable regions surrounding the binding site for host sialic acid receptor and inhibited viral attachment to cell surfaces. Owing to continual genetic evolution of this region of haemagglutinin, however, antibodies specific for the head region usually bind to a restricted subset of viral strains. However, in 2008–2009, two important papers74,75 were published that determined the structure of two different broadly neutralizing antibodies specific for the HA2 subunit. These antibodies were isolated by phage display from B cells of people vaccinated against influenza74,76 or from a synthetic library75. These two antibodies prevent the structural rearrangements of haemagglutinin that are necessary for the fusion of the viral envelope with internal cell membranes. Because this portion of the haemagglutinin stem region is highly conserved across influenza virus strains, these antibodies neutralized most strains in the H1, H5 and H9 clade. Currently there is a substantial effort in the influenza vaccine community to preferentially target these epitopes72.

a | The figure shows a space-filling model of a haemagglutinin trimer from the 2009 pandemic H1N1 influenza virus strain (the structural data were obtained from the Protein Data Bank (PDB ID: 3LZG))77. The HA1 and HA2 subunits of one trimer — as well as how they contribute to the haemagglutinin globular head region versus the more conserved haemagglutinin stalk region — are indicated. b | The figure shows a top-down view of a space-filling model of HIV gp120 (PDB ID: 2NY6), and indicates the crucial CD4 binding site, as determined by the Kwong group92.
The discovery of these broadly neutralizing antibodies prompted the search for and characterization of additional broadly neutralizing human monoclonal antibodies specific for influenza virus. Using B cells from individuals that had received the annual seasonal trivalent influenza vaccine containing H1, H3 and B strains, EBV-immortalized memory B cells with broadly neutralizing specificities were identified by screening for monoclonal antibodies that could neutralize the H5 strain22. By screening for neutralization of a viral strain not present in the vaccine, the investigators could identify B cells with antibodies able to bind to regions that are conserved between divergent strains. In this way, 20 independent human monoclonal antibodies that bound to viruses of the H1 and H5 clades were isolated, as well as a few antibodies that recognized H9 clade viruses. All but one of these monoclonal antibodies bound to the stem portion of haemagglutinin, and the one head-binding antibody had a more restricted virus strain neutralization capability. Another study used the novel approach of culturing single plasma cells that were isolated a week after either influenza virus infection or administration of the trivalent vaccine9. The authors then screened the single-cell culture supernatants for the presence of immunoglobulins capable of binding to both group 1 and group 2 haemagglutinin. Although the low antibody concentration in the supernatant obtained using this method may preclude the detection of some low-affinity antibodies, sufficient amounts of antibody are generated from high-affinity clones to screen multiple antigens. Using this approach, they were able to identify one antibody that bound to both group 1 and group 2 influenza virus strains9. However, the natural emergence of these very broadly neutralizing antibodies appears to be rare, as the antibody that bound to both groups was the only one obtained from among 104,000 plasma cells from 8 different donors previously selected for high heterosubtypic serum antibody titres. Furthermore, this same antibody could not be identified when 20,000 immortalized memory B cells from the same donor were screened9.
The emergence of the 2009 pandemic H1N1 strain created an interesting situation in which the ability to clone and characterize the specificity of plasmablasts from peripheral blood could be applied. The pandemic H1N1 strain arose from a reassortment of swine and human viral components in which the haemagglutinin and neuraminidase proteins were derived from the swine influenza virus. This H1N1 strain most closely resembled the 1918 pandemic H1N1 virus, with ∼20% divergence between the two virus strains at major antigenic sites on the haemagglutinin head. By contrast, there was 50% divergence between the 2009 pandemic H1N1 strain and the most recent seasonal vaccine strain of H1N1 influenza (namely A/Brisbane/10/2007)77. Not surprisingly, older individuals, who were more likely to have been exposed to 1918-like strains, had fewer incidents of 2009 pandemic H1N1 influenza infection than younger people. What soon became apparent, however, in analysing the plasmablast response in relatively young individuals exposed to the pandemic H1N1 strain, was that a much higher percentage of responding cells were broadly reactive across the H1 and H5 viral strains. In contrast to the response to pre-pandemic seasonal H1N1 strains, in which antibodies typically bound to the antigenically divergent globular head, antibodies induced by the 2009 pandemic strain bound to the conserved region on the haemagglutinin stem64. In addition to the broad reactivity of these antibodies, the immunoglobulin variable region genes from the pandemic H1N1-induced plasmablasts were highly mutated. This indicates that these plasmablasts probably arose from memory B cells with heterosubtypic specificities, which are known to exist at low frequencies owing to past exposure to seasonal H1N1 strains22. Taken together, these data indicate that exposure to an antigenically novel strain of influenza virus elicits an antibody response that is biased towards antibodies specific for a more conserved region of haemagglutinin, namely the stem region. This observation provided an important proof of principle that a broadly protective or even a pan-protective antibody response to influenza virus could indeed be elicited with the right immunogen. Subsequent studies have now demonstrated that a similar response can be induced in humans in response to the pandemic H1N1 strain vaccine78,79.
HIV. HIV has been intensely studied since it was first isolated in the 1980s, but an effective vaccine has remained elusive owing to the T cell tropism and the immune evasion capacity of the virus80. In particular, the development of broadly neutralizing antibodies to the viral envelope — which contains spikes that are composed of a surface gp120 trimer and a transmembrane gp41 trimer (Fig. 3b) — is impeded by the high variability of the exposed epitopes and the structural inaccessibility of conserved regions owing in part to heavy glycan shielding81. Because of the relative difficulty of generating human monoclonal antibodies and the rarity of broadly neutralizing antibodies in infected individuals, until recently only a few naturally occurring broadly neutralizing antibodies specific for conserved regions of HIV had been described82,83. Since 2009, however, a rapid succession of papers describing newly isolated broadly neutralizing antibodies from HIV-infected patients has indicated that potent broadly neutralizing antibodies can be naturally generated. This has provided important insights into the crucial epitopes that might facilitate the development of an HIV vaccine and the ways in which these epitopes might be targeted.
The success of isolating broadly neutralizing antibodies generated in response to human HIV infection in recent years has been due to careful selection of infected donors with high serum titres of broadly neutralizing antibodies and due to targeted approaches to isolate large quantities of HIV-specific neutralizing memory B cells. The first high-throughput attempt began by screening serum from ∼1,800 HIV-1-infected donors for high neutralizing titres84. Next, Simek et al. chose one donor from whom they activated and differentiated ∼30,000 IgG+CD19+ memory B cells into antibody-secreting cells. They then screened culture supernatants for antibodies capable of binding to the envelope proteins gp120 and gp41 and for neutralizing activity10. In this screen, two broadly neutralizing monoclonal antibodies were isolated that had higher neutralizing activities than any of the other broadly neutralizing antibodies isolated thus far. The binding of the two broadly neutralizing antibodies mapped to a conserved region of the HIV gp120 subunit containing variable loop residues and N-linked glycans85. Screening memory B cells from another four infected donors yielded several additional broadly neutralizing antibodies with even more potent broadly neutralizing capabilities that also bound to variable loop epitopes27. Corti et al. screened EBV-immortalized memory B cells from 21 HIV-infected donors with neutralizing serum antibodies26. The efficacy of EBV immortalization was lower for B cells from HIV-positive patients than for B cells from healthy donors, but 58 B cell clones were isolated, one of which had potent and broad neutralizing capability.
Wu et al. used a highly targeted flow cytometry-based approach to isolate broadly neutralizing antibodies from memory B cells of infected individuals. Based on the epitope that is bound by the well-characterized broadly neutralizing antibody b12, which binds to the CD4 binding site on gp120, a stabilized core of gp120 was constructed that contained the major contact sites for CD4, but from which other antigenic regions to which non-neutralizing antibodies bind were removed55. Using this resurfaced protein (termed RSC3) and the same protein with a point mutation in the CD4 binding site (ΔRSC3) as bait, the investigators sorted 29 memory B cells that had specificity for RSC3 but not for ΔRSC3 from a total of 25 million peripheral blood mononuclear cells from one donor whose serum had been shown to bind to RSC3 during pre-screening. From these individually sorted B cells, they directly amplified and cloned the immunoglobulin heavy and light chain genes and made human monoclonal antibodies. Three of these human monoclonal antibodies (VRC01, VRC02 and VRC03) bound to RSC3, two of which were clonally related. These clones bound gp120 with a relatively high affinity and neutralized an impressive breadth of HIV isolates. The authors later used the same approach with two more donors and identified more potent broadly neutralizing antibodies that likewise bound to the CD4 binding site on gp120 (Ref. 56).
Scheid et al. used a similar approach to screen memory B cells for HIV binding but, instead of targeting B cells that specifically bound the CD4 binding site, they used a soluble form of the complete trimerized Env spike (gp140) as bait for flow cytometry to screen memory B cells from six patients who had been pre-screened for broadly neutralizing antibodies in their blood52. This was a highly efficient method, as 86% of the B cells from which monoclonal antibodies were produced were specific for HIV. From the 433 HIV-binding human monoclonal antibodies that were made, 134 were clonally distinct, and 65 of these were neutralizing against a narrow range of HIV isolates, binding to a spectrum of gp120 epitopes. This study demonstrated the wide range of neutralizing epitopes that exist when considering specific HIV isolates. When these human monoclonal antibodies were used as a cocktail at high concentrations, they together neutralized a broad array of HIV isolates, although no single monoclonal antibody had the same neutralizing breadth as the monoclonal antibodies that have been previously described. However, by altering the primers used to amplify the immunoglobulin genes to reproduce the high mutation level of HIV-specific broadly neutralizing antibodies, and by using a more-targeted probe composed of the gp120 core glycoprotein stabilized in the CD4-bound conformation but lacking variable loops 1–3, six clonally distinct broadly neutralizing antibodies were identified53. One of the monoclonal antibodies (NIH45-46) was a more potent variant of the VRC01 broadly neutralizing antibody isolated from the same patient using RSC3 as bait56.
In the case of HIV, the field has progressed further than simply isolating and characterizing naturally produced broadly neutralizing antibodies. The groups mentioned above have gone on to comprehensively study, both at the DNA sequence and the protein structure level, how these antibodies were generated and which features are essential for their potency. For this, Scheid et al. used primers specific for the immunoglobulin variable heavy chain genes of the broadly neutralizing antibody to amplify large numbers of additional cDNA transcripts from related clones from long-lived bone marrow plasma cells of the same HIV-infected donors53. This both provided information on the variability and magnitude of clonal variants and tested for the presence of B cells with certain specificities in different B cell effector populations. Wu et al. performed next-generation sequencing on memory B cell cDNA and, by focusing on the same immunoglobulin variable heavy chain genes, obtained thousands of sequences with over 75% identity to the original B cell immunoglobulin genes sorted by flow cytometry56. By making monoclonal antibodies from some of these variants and examining their functional characteristics, a remarkable convergence by affinity maturation to conserved residues that are important in neutralizing a wide range of HIV variants was identified53,56. At the protein structure level, Diskin et al. compared the structures of VRC01 and NIH45-46 in complex with the HIV gp120, and this led to the in silico design of an antibody with an even greater breadth of neutralizing activity than either of the original forms86.
All together, these elegant studies serve to illustrate the advantages and disadvantages of the different strategies used to isolate naturally produced human monoclonal antibodies. By using a very specific probe that is designed based on the lessons learnt from previously isolated neutralizing and non-neutralizing monoclonal antibodies, Wu et al. were able to isolate broadly neutralizing antibodies that recognized a highly conserved epitope that is crucial for viral infection55,56. Walker et al. chose to screen for the neutralizing capacity of memory B cells, and not to base their selection on the binding of antibodies to specific regions of the HIV spike10,27. In this way, they obtained broadly neutralizing antibodies that recognized conserved glycan–protein quarternary structures in variable loop regions of gp120 that were thought to be poor targets for broadly neutralizing antibodies owing to their mostly variable residues27,85,87. By using binding to the HIV spike as the only selection criterion, Scheid et al. and Mouquet et al. did not focus on identifying only the most broadly neutralizing B cells, but instead produced information on the diversity of the epitopes targeted by neutralizing B cells in response to HIV52,54. This widens the list of epitopes that need to be considered when designing vaccine immunogens. It also provides a large dataset from which to glean the common features of HIV-specific antibodies and sets the groundwork for the comparison of B cell responses between individuals with different clinical outcomes.
The challenge: inducing neutralizing antibodies
As we continue to focus on the best ways to identify and characterize naturally produced human monoclonal antibodies, the ongoing challenge is to take advantage of the information we learn. Although these human monoclonal antibodies might certainly have exciting potential as therapeutics, the highest impact on public health would be achieved by the design of vaccines that naturally elicit antibodies that bind to key epitopes. As reviewed in detail elsewhere72,82,88, several general approaches are being considered that exemplify how the B cell response might be targeted by new vaccines.
One of the biggest challenges is to increase the breadth of protection provided by a vaccine to neutralize a large span of evolving viral strains. However, the isolation of neutralizing human monoclonal antibodies from some individuals does not mean that we can readily induce similar antibodies in all vaccinated individuals. The immune response of individuals can be dramatically affected by various factors, including immune and vaccine history, genetic background, geographical region and ethnicity, diet, concurrent infections and age or other factors that cause them to be immunocompromised. For example, although some individuals clearly responded to the 2009 pandemic H1N1 infection or vaccination with a particularly broad-spectrum response, others in these studies had highly specific responses to the pandemic H1N1 strain64,78,79. Because the more broadly protective response was probably elicited from memory B cells specific for divergent influenza strains, it is possible that the people with highly targeted responses had few pre-existing memory B cells to influenza virus. In the case of HIV, it has been estimated that 20–30% of infected individuals have broadly neutralizing antibodies89,90. An open question is whether few people develop these antibodies because of differences in the pre-existing B cell repertoire or because of inefficient B cell activation. HIV-specific broadly neutralizing antibodies are for the most part highly mutated and often polyreactive53,91. Therefore the generation of these broadly neutralizing antibodies may require extensive antigen exposure and multiple rounds of hypermutation and affinity maturation. Tolerance mechanisms in some individuals may also prevent these polyreactive B cells from reaching maturity. This will be an important point to determine as vaccination approaches against HIV are explored.
In any event, a key first step is identifying the relevant epitopes that need to be targeted and understanding in detail the immune processes required for the natural production of neutralizing antibodies. Use of the various approaches discussed here to more efficiently clone human monoclonal antibodies should continue to provide crucial insights into how we can finally develop vaccines to immunize against pathogens that have thus far evaded our efforts.
References
Casadevall, A., Dadachova, E. & Pirofski, L. A. Passive antibody therapy for infectious diseases. Nature Rev. Microbiol. 2, 695–703 (2004).
Kohler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
Winter, G. & Harris, W. J. Humanized antibodies. Immunol. Today 14, 243–246 (1993).
Carter, P. J. Potent antibody therapeutics by design. Nature Rev. Immunol. 6, 343–357 (2006).
Chan, A. C. & Carter, P. J. Therapeutic antibodies for autoimmunity and inflammation. Nature Rev. Immunol. 10, 301–316 (2010).
Weiner, L. M., Surana, R. & Wang, S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nature Rev. Immunol. 10, 317–327 (2010).
Bradbury, A. R., Sidhu, S., Dubel, S. & McCafferty, J. Beyond natural antibodies: the power of in vitro display technologies. Nature Biotech. 29, 245–254 (2011).
Jin, A. et al. A rapid and efficient single-cell manipulation method for screening antigen-specific antibody-secreting cells from human peripheral blood. Nature Med. 15, 1088–1092 (2009).
Corti, D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011). This study describes the isolation of an antibody that broadly neutralizes both group 1 and group 2 influenza virus strains.
Walker, L. M. et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289 (2009). This study used a very targeted high-throughput method to detect and isolate HIV-specific broadly neutralizing antibodies.
Benckert, J. et al. The majority of intestinal IgA+ and IgG+ plasmablasts in the human gut are antigen-specific. J. Clin. Invest. 121, 1946–1955 (2011).
Marasco, W. A. & Sui, J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nature Biotech. 25, 1421–1434 (2007).
Marks, J. D. et al. By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology 10, 779–783 (1992).
Steinitz, M., Klein, G., Koskimies, S. & Makel, O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature 269, 420–422 (1977).
Burioni, R. et al. Monoclonal antibodies isolated from human B cells neutralize a broad range of H1 subtype influenza A viruses including swine-origin influenza virus (S-OIV). Virology 399, 144–152 (2010).
Habersetzer, F. et al. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology 249, 32–41 (1998).
Atlaw, T., Kozbor, D. & Roder, J. C. Human monoclonal antibodies against Mycobacterium leprae. Infect. Immun. 49, 104–110 (1985).
Cole, S. P., Campling, B. G., Atlaw, T., Kozbor, D. & Roder, J. C. Human monoclonal antibodies. Mol. Cell. Biochem. 62, 109–120 (1984).
Buchacher, A. et al. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res. Hum. Retroviruses 10, 359–369 (1994).
Traggiai, E. et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nature Med. 10, 871–875 (2004). This paper describes the first published improved method for the EBV-mediated transformation of memory B cells, which increased the efficiency of B cell immortalization to a level that has allowed the isolation of naturally generated monoclonal antibodies in many diseases.
Krause, J. C. et al. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J. Virol. 85, 10905–10908 (2011).
Corti, D. et al. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J. Clin. Invest. 120, 1663–1673 (2010).
Krause, J. C. et al. Human monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza viruses. J. Virol. 86, 6334–6340 (2012).
Yu, X. et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature 455, 532–536 (2008).
Bonsignori, M. et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 85, 9998–10009 (2011).
Corti, D. et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS ONE 5, e8805 (2010).
Walker, L. M. et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011).
Macagno, A. et al. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 84, 1005–1013 (2010).
Beltramello, M. et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8, 271–283 (2010).
Dejnirattisai, W. et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science 328, 745–748 (2010).
Smith, S. A. et al. Persistence of circulating memory B cell clones with potential for dengue virus disease enhancement for decades following infection. J. Virol. 86, 2665–2675 (2012).
Schieffelin, J. S. et al. Neutralizing and non-neutralizing monoclonal antibodies against dengue virus E protein derived from a naturally infected patient. Virol. J. 7, 28 (2010).
de Alwis, R. et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl Acad. Sci. USA 109, 7439–7444 (2012).
de Alwis, R. et al. In-depth analysis of the antibody response of individuals exposed to primary dengue virus infection. PLoS Negl. Trop. Dis. 5, e1188 (2011).
Warter, L. et al. Chikungunya virus envelope-specific human monoclonal antibodies with broad neutralization potency. J. Immunol. 186, 3258–3264 (2011).
Collarini, E. J. et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J. Immunol. 183, 6338–6345 (2009).
Yu, X., McGraw, P. A., House, F. S. & Crowe, J. E. Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J. Immunol. Methods 336, 142–151 (2008).
Kwakkenbos, M. J. et al. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nature Med. 16, 123–128 (2010).
Wardemann, H. et al. Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003).
Wardemann, H. & Nussenzweig, M. C. B-cell self-tolerance in humans. Adv. Immunol. 95, 83–110 (2007).
Scheid, J. F. et al. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl Acad. Sci. USA 108, 18044–18048 (2011).
Meffre, E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann. NY Acad. Sci. 1246, 1–10 (2011).
Isnardi, I. et al. Complement receptor 2/CD21− human naive B cells contain mostly autoreactive unresponsive clones. Blood 115, 5026–5036 (2010).
Duty, J. A. et al. Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J. Exp. Med. 206, 139–151 (2009).
Smith, K. et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nature Protoc. 4, 372–384 (2009).
Casali, P., Inghirami, G., Nakamura, M., Davies, T. F. & Notkins, A. L. Human monoclonals from antigen-specific selection of B lymphocytes and transformation by EBV. Science 234, 476–479 (1986).
Weitkamp, J. H. et al. Generation of recombinant human monoclonal antibodies to rotavirus from single antigen-specific B cells selected with fluorescent virus-like particles. J. Immunol. Methods 275, 223–237 (2003).
Tiller, T. et al. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124 (2008).
Scheid, J. F. et al. A method for identification of HIV gp140 binding memory B cells in human blood. J. Immunol. Methods 343, 65–67 (2009).
Di Niro, R. et al. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J. Immunol. 185, 5377–5383 (2010).
Di Niro, R. et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nature Med. 18, 441–445 (2012).
Scheid, J. F. et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458, 636–640 (2009).
Scheid, J. F. et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637 (2011). This study describes the comprehensive isolation and characterization of broadly neutralizing HIV-specific antibody clonal families.
Mouquet, H. et al. Memory B cell antibodies to HIV-1 gp140 cloned from individuals infected with clade A and B viruses. PLoS ONE 6, e24078 (2011).
Wu, X. et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 (2010). This paper describes the design of a probe containing conserved regions of gp120 to specifically detect and isolate rare broadly neutralizing antibodies specific for HIV using flow cytometry.
Wu, X. et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333, 1593–1602 (2011).
Whittle, J. R. et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl Acad. Sci. USA 108, 14216–14221 (2011).
Poulsen, T. R., Meijer, P. J., Jensen, A., Nielsen, L. S. & Andersen, P. S. Kinetic, affinity, and diversity limits of human polyclonal antibody responses against tetanus toxoid. J. Immunol. 179, 3841–3850 (2007).
Liao, H. X. et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J. Virol. Methods 158, 171–179 (2009).
Bernasconi, N. L., Traggiai, E. & Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 298, 2199–2202 (2002).
Brokstad, K. A., Cox, R. J., Olofsson, J., Jonsson, R. & Haaheim, L. R. Parenteral influenza vaccination induces a rapid systemic and local immune response. J. Infect. Dis. 171, 198–203 (1995).
Sasaki, S. et al. Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J. Virol. 81, 215–228 (2007).
Wrammert, J. et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008). In this study, the authors identified and described a method for using short-lived plasmablasts generated in response to influenza vaccination as a good source of naturally produced monoclonal antibodies.
Wrammert, J. et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208, 181–193 (2011). Using human monoclonal antibody technologies, this study showed that with the appropriate immunogen an antibody response dominated by broadly neutralizing antibodies could indeed be induced in humans.
Meijer, P. J. et al. Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J. Mol. Biol. 358, 764–772 (2006).
Poulsen, T. R., Jensen, A., Haurum, J. S. & Andersen, P. S. Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J. Immunol. 187, 4229–4235 (2011).
Sasaki, S. et al. Limited efficacy of inactivated influenza vaccine in elderly individuals is associated with decreased production of vaccine-specific antibodies. J. Clin. Invest. 121, 3109–3119 (2011).
Moody, M. A. et al. H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination. PLoS ONE 6, e25797 (2011).
Reddy, S. T. & Georgiou, G. Systems analysis of adaptive immunity by utilization of high-throughput technologies. Curr. Opin. Biotechnol. 22, 584–589 (2011).
Krause, J. C. et al. Epitope-specific human influenza antibody repertoires diversify by B cell intraclonal sequence divergence and interclonal convergence. J. Immunol. 187, 3704–3711 (2011).
Cheung, W. C. et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nature Biotech. 30, 447–452 (2012).
Kaur, K., Sullivan, M. & Wilson, P. C. Targeting B cell responses in universal influenza vaccine design. Trends Immunol. 32, 524–531 (2011).
Okuno, Y., Isegawa, Y., Sasao, F. & Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 67, 2552–2558 (1993).
Ekiert, D. C. et al. Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251 (2009).
Sui, J. et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nature Struct. Mol. Biol. 16, 265–273 (2009). References 74 and 75 are the first papers to determine the conserved epitope and crystal structure of a broadly neutralizing influenza-specific antibody in complex with viral haemagglutinin.
Throsby, M. et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 3, e3942 (2008).
Xu, R. et al. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328, 357–360 (2010).
Thomson, C. A. et al. Pandemic H1N1 influenza infection and vaccination in humans induces cross-protective antibodies that target the hemagglutinin stem. Front. Immunol. 3, 1–19 (2012).
Li, G.-M. et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc. Natl Acad. Sci. USA 21 May 2012 (doi:10.1073/pnas.1118979109).
McMichael, A. J., Borrow, P., Tomaras, G. D., Goonetilleke, N. & Haynes, B. F. The immune response during acute HIV-1 infection: clues for vaccine development. Nature Rev. Immunol. 10, 11–23 (2010).
Pantophlet, R. & Burton, D. R. GP120: target for neutralizing HIV-1 antibodies. Annu. Rev. Immunol. 24, 739–769 (2006).
Moir, S., Malaspina, A. & Fauci, A. S. Prospects for an HIV vaccine: leading B cells down the right path. Nature Struct. Mol. Biol. 18, 1317–1321 (2011).
Kwong, P. D. & Wilson, I. A. HIV-1 and influenza antibodies: seeing antigens in new ways. Nature Immunol. 10, 573–578 (2009).
Simek, M. D. et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J. Virol. 83, 7337–7348 (2009).
McLellan, J. S. et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343 (2011).
Diskin, R. et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334, 1289–1293 (2011). By studying the structure of broadly neutralizing antibodies bound to gp120 isolated from an HIV-infected individual, the authors of this study were able to design an improved antibody in silico.
Pejchal, R. et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103 (2011).
Haynes, B. F., Kelsoe, G., Harrison, S. C. & Kepler, T. B. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nature Biotech. 30, 423–433 (2012).
Doria-Rose, N. A. et al. Frequency and phenotype of human immunodeficiency virus envelope-specific B cells from patients with broadly cross-neutralizing antibodies. J. Virol. 83, 188–199 (2009).
Sather, D. N. et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J. Virol. 83, 757–769 (2009).
Mouquet, H. et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595 (2010).
Zhou, T. et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445, 732–737 (2007).
Simmons, C. P. et al. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 4, e178 (2007).
Hu, H. et al. A human antibody recognizing a conserved epitope of H5 hemagglutinin broadly neutralizes highly pathogenic avian influenza H5N1 viruses. J. Virol. 86, 2978–2989 (2012).
Kashyap, A. K. et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl Acad. Sci. USA 105, 5986–5991 (2008).
Tian, C. et al. Immunodominance of the VH1-46 antibody gene segment in the primary repertoire of human rotavirus-specific B cells is reduced in the memory compartment through somatic mutation of nondominant clones. J. Immunol. 180, 3279–3288 (2008).
Acknowledgements
K. Kaur and C. Dunand provided helpful suggestions. This work was funded in part by grants from the US National Institute of Allergy and Infectious Diseases, National Institutes of Health: 5U54AI057158-08 (to P.C.W.), 5U19AI057266-08 (to P.C.W.), 5U19AI082724-03 (to P.C.W.), 1U19AI090023-01 (to P.C.W.) and F32AI930872 (to S.F.A.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Hybridoma technology
-
Fusion of activated, antibody-secreting primary B cells with an immortalized myeloma cell line to produce long-living cell lines expressing antibodies of a single specificity.
- Phage display libraries
-
Pools of bacteriophage virions, each expressing a unique protein variant (such as immunoglobulin fragments) on the virion exterior.
- AID
-
(Activation-induced cytidine deaminase). A cytosine deaminase that catalyses a pivotal step in antibody gene-diversification reactions.
- Reverse transcription PCR
-
(RT-PCR). A type of PCR in which RNA is first converted into double-stranded DNA, which is then amplified.
- Next-generation sequencing
-
A massively parallel high-throughput non-Sanger method of sequencing.
Rights and permissions
About this article
Cite this article
Wilson, P., Andrews, S. Tools to therapeutically harness the human antibody response. Nat Rev Immunol 12, 709–719 (2012). https://doi.org/10.1038/nri3285
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3285
This article is cited by
-
Development and application of classical swine fever virus monoclonal antibodies derived from single B cells
Veterinary Research (2023)
-
Monoclonal antibodies from humans with Mycobacterium tuberculosis exposure or latent infection recognize distinct arabinomannan epitopes
Communications Biology (2021)
-
Recent advances in therapeutic applications of neutralizing antibodies for virus infections: an overview
Immunologic Research (2020)
-
Computational B-cell epitope identification and production of neutralizing murine antibodies against Atroxlysin-I
Scientific Reports (2018)
-
Single human B cell-derived monoclonal anti-Candida antibodies enhance phagocytosis and protect against disseminated candidiasis
Nature Communications (2018)
