
Key Points
-
C-type lectin receptors (CLRs) are efficient pattern-recognition receptors (PRRs) that interact with pathogens via carbohydrate structures, leading to enhanced antigen presentation as well as modulation of T helper cell (TH cell) differentiation, collectively inducing pathogen-tailored adaptive immune responses.
-
Crosstalk between innate signalling by CLRs and other PRRs, as well as other receptors such as type I interferon receptor (IFNAR), enhances the diversity of TH cell responses to pathogens.
-
Engagement of the CLR dectin 1 by fungi induces various intracellular signalling pathways that cooperate to induce efficient TH1 and TH17 cell responses, which are crucial for antifungal immunity.
-
DC-SIGN (DC-specific ICAM3-grabbing non-integrin), another CLR, induces specific signalling cascades for distinct pathogens, according to the carbohydrate ligand recognized. The recognition of mannose on intracellular pathogens such as fungi, viruses and mycobacteria leads to protective TH1 cell responses when triggered together with Toll-like receptor (TLR) signalling. By contrast, parasitic fucose-containing ligands induce both TH2 and T follicular helper (TFH) cell responses (which are necessary for humoral immunity) during crosstalk of DC-SIGN signalling with TLR and IFNAR signalling.
-
CLR triggering also contributes to pathogenic disorders; dectin 2 engagement by allergens originating from fungi or house dust mites leads to allergic TH2 cell responses, whereas triggering of macrophage-inducible C-type lectin receptor (MINCLE) by pathogenic Fonsecaea spp. fungi represses TH1 cell responses, thereby promoting fungal dissemination.
-
CLRs have previously been used in vaccine strategies for their antigen-processing capacity. Our advanced knowledge of the signalling pathways by which CLRs direct adaptive immunity now provides a powerful tool to add a novel level of sophistication to the design of vaccines. In particular, the very specific TFH cell responses that are induced by CLRs such as DC-SIGN and CLEC9A could greatly improve the generation of broadly neutralizing antibodies against viruses such as HIV-1.
Abstract
Pathogen recognition by C-type lectin receptors (CLRs) expressed by dendritic cells is important not only for antigen presentation, but also for the induction of appropriate adaptive immune responses via T helper (TH) cell differentiation. CLRs act either by themselves or in cooperation with other receptors, such as other CLRs, Toll-like receptors and interferon receptors, to induce signalling pathways that trigger specialized cytokine programmes for polarization of TH cell differentiation. In this Review, we discuss how triggering of the prototypical CLRs leads to distinct pathogen-tailored TH cell responses and how we can harness our expanding knowledge for vaccine design and the treatment of inflammatory and malignant diseases.
Similar content being viewed by others
Main
Efficient T cell responses require differentiation of CD4+ T cells into different T helper (TH) cell subsets, depending on the type of infection. These TH cell subsets each have specific roles in the defence against pathogens by helping other innate and adaptive immune cells to mount effective responses against the infectious agent. Antigen-presenting cells and in particular dendritic cells (DCs) orchestrate TH cell differentiation by stimulating T cell receptors and creating a specialized cytokine environment. The prime determinant that controls TH cell differentiation is pathogen recognition by DCs, which translates the nature of the invading pathogen into a gene-transcriptional programme that drives the appropriate TH cell response1.
Several different TH cell subsets have been identified that each have specific functions in adaptive immunity (Fig. 1). TH1 and TH2 cell responses are important in host defence against intracellular and extracellular pathogens, respectively2,3. Interleukin-12p70 (IL-12p70) drives TH1 cell differentiation via the induction of the transcription factor T-bet (also known as TBX21)4. The underlying mechanisms of TH2 cell differentiation are not completely understood, but this process is reciprocally entwined with TH1 cell differentiation, as inhibition of IL-12p70 secretion from DCs facilitates TH2 cell differentiation, whereas induction of the co-stimulatory molecule OX40L (also known as TNFSF4) primes DCs towards TH2 cell polarization5,6.

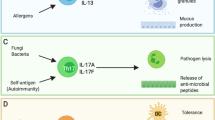
T helper 1 cell (TH1 cell) responses are required for defence against intracellular pathogens such as fungi, viruses and mycobacteria, and are induced by dendritic cells (DCs) that secrete interleukin-12p70 (IL-12p70). T-bet is the master regulator of TH1 cell differentiation and is regulated by signal transducer and activator of transcription 4 (STAT4), which is induced by IL-12p70. TH2 cell responses defend against extracellular pathogens by activating eosinophils and basophils and by inducing antibody isotype switching to IgE in B cells3. TH2 cells are characterized by the secretion of IL-4, IL-5 and IL-13 and by the expression of the transcription factor GATA3, which is induced by STAT6 signalling. TH17 cell responses are crucial in defence against fungi through the secretion of IL-17A, IL-17F and IL-22, which recruit neutrophils to sites of infection7. The master regulator, RORγt, is induced by STAT3 signalling via simultaneous IL-1β and IL-6 stimulation (humans) and additionally transforming growth factor-β (TGFβ; mice), whereas IL-23 plays an important part in the maintenance of TH17 cell commitment. T follicular helper cell (TFH cell) responses, induced by DC-derived signals such as IL-6 and IL-27, are essential for the establishment of protective long-term humoral immunity against pathogens via the generation of high-affinity antibodies, which are induced through the production of IL-21. The autocrine action of IL-21 regulates B cell lymphoma 6 (BCL-6) expression. BCL-6, induced by STAT3 signalling, acts as the master regulator of TFH cell differentiation, including the induction of IL-21 and CXC-chemokine receptor 5 (CXCR5), which is important for TFH cell homing to B cell zones. CCL, CC-chemokine ligand; GC, germinal centre; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFNγ, interferon-γ; NK cell, natural killer cell; PRR, pattern-recognition receptor; TNF, tumour necrosis factor.
TH17 cell responses are crucial for defence against fungi7, and the expression of the transcription factor RORγt, the master regulator of TH17 cells, is induced via IL-1β and IL-6 stimulation in humans and via transforming growth factor-β (TGFβ) stimulation in mice8, whereas IL-23 plays an important part in the maintenance of TH17 cell commitment9.
T follicular helper (TFH) cell responses establish protective long-term humoral immunity against pathogens10. Multiple signals act in concert to initiate TFH cell differentiation at the time of DC priming; in particular, IL-6 and IL-21 have important roles in vivo in the generation and maintenance of TFH cells and in the formation of germinal centres in mice10. Recently, it was shown that IL-27 is crucial for inducing mouse and human TFH cell differentiation11,12.
Several other TH cell subsets, such as TH9 cells and TH22 cells13,14, help in defence against pathogens, whereas regulatory T cells are important for limiting immune responses, thereby preventing autoimmune and other deleterious immune responses15.
In this Review, we focus on the induction of TH cell responses by C-type lectin receptors (CLRs) that function as pattern-recognition receptors (PRRs) on DCs. CLRs constitute a large superfamily of proteins that are defined by the presence of at least one C-type lectin recognition domain16. The entire CLR family recognizes a diverse range of carbohydrate structures, such as mannose, fucose, sialic acid and β-glucan, and the exact carbohydrate (or carbohydrates) recognized by a CLR dictates which pathogens are bound by that receptor17. CLRs are expressed by myeloid cells and in particular by macrophages and DC subsets, but some CLRs, such as dectin 1 (also known as CLEC7A), are also expressed by neutrophils, B cells and keratinocytes18,19. The differential expression of CLRs by DC subsets dictates how these subsets react to pathogens and underlies their specialized functions; thus, it is not surprising that other CLRs, such as langerin (also known as CLEC4K) and CLEC9A, have a restricted expression pattern20,21.
Here, we focus on the current understanding of CLRs that sense invading pathogens and subsequently induce specialized cytokine expression profiles. Specifically, we integrate knowledge of these pathways with the role of particular TH cell responses in infection, and discuss how we can harness this information in the design of vaccines for infectious diseases, autoimmune diseases and malignant disorders.
CLRs and TH cell differentiation
CLRs differ from most other PRRs in that pathogen recognition via high-affinity and high-avidity interactions leads to pathogen uptake and degradation in the antigen-processing machinery, as well as the activation of innate signalling pathways that induce or modulate innate and adaptive immune responses17. Many CLRs can sense different classes of pathogen, as well as malignant tumour cells, by their glycosylation pattern — the so-called carbohydrate fingerprint17. Bacteria, parasites and fungi have glycosylation structures that are distinct from host carbohydrates. Interestingly, even though viruses hijack the glycosylation machinery of host cells, their glycosylated envelope proteins differ from endogenous proteins in the amount and the composition of glycosylation: structures with a high mannose content are more prevalent in viral envelope glycoproteins than complex and hybrid glycans, which are common on host cells22. Malignant host cells also exhibit a distinct activation and/or expression of glycosyltransferases, often resulting in tumour-specific glycosylation patterns23. This makes CLRs well equipped to distinguish between different pathogens, to recognize tumour cells and to orchestrate the appropriate adaptive immune responses.
Dectin 1 induces TH1 and TH17 cell responses
A prototypic CLR is dectin 1, which is crucial for defence against fungi, as underscored by the finding that individuals with a defective dectin 1 are more susceptible to Candida spp. infections24. Although dectin 1-deficient mice are also less resistant to various fungal infections than wild-type mice25,26, their level of susceptibility is dependent on the infecting fungal strain, owing to adaptation of fungal cell wall composition in vivo27. As shown in mice, dectin 1 is also important for maintaining microbiota homeostasis in the colon by controlling the growth of resident fungi28.
Dectin 1 recognizes β-glucans that are exposed at the budding scars of pathogenic and opportunistic fungi (Table 1). It is expressed by Langerhans cells (LCs) and by different myeloid DCs, including inflammatory DCs, CD1c+ DCs and CD141+ DCs, which places this receptor in an ideal position to detect invasive fungi17. LCs are important for the induction of TH17 cell responses to cutaneous fungal infections17,29, whereas subepithelial DCs induce both TH17 and TH1 cell responses29,30,31, which are important for defence against systemic invasive fungal infections29.
Dectin 1-mediated signalling is activated only by particulate β-glucans, which cluster the receptor in synapse-like structures32. Early studies showed that dectin 1 collaborates with signalling by Toll-like receptor 2 (TLR2) to enhance cytokine responses in macrophages33. However, dectin 1 is now known to also have a direct role in adaptive immunity by triggering several distinct signalling pathways that cooperate to shape TH cell responses in response to fungal infections30,31,34,35.
Ligand binding by dectin 1 and subsequent receptor clustering triggers phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM)-like sequence within the cytoplasmic domains of the dectin 1 receptors, leading to the consecutive recruitment of SHP2 (also known as PTPN11) and the tyrosine kinase SYK, followed by the formation of the CARD9–BCL-10–MALT1 (caspase recruitment domain-containing protein 9–B cell lymphoma 10–mucosa-associated lymphoid tissue lymphoma translocation protein 1) scaffold complex30,31,34,35,36 (Fig. 2). This scaffold initiates a signalling cascade that leads to activation of both the canonical NF-κB pathway (nuclear factor-κB pathway) and the non-canonical NF-κB pathway; that is, the activation of canonical p65–p50, canonical REL–p50 and non-canonical RELB–p52 transcriptional complexes30.

The recognition of fungal β-glucans such as those of Candida albicans by dectin 1 induces phosphorylation (P) of its immunoreceptor tyrosine-based activation motif (ITAM)-like sequence by an unidentified SRC kinase (not shown). Subsequently, SHP2 binds to the ITAM-like sequence via its N-SH2 domain, and phosphorylation of the ITAM- like sequence of SHP2 by either a SRC kinase or the tyrosine kinase SYK facilitates the recruitment of SYK to the receptor complex36. Dimerization of dectin 1 brings two SYK molecules together, leading to trans-activation of SYK, which induces the assembly of a CARD9–BCL-10–MALT1 (caspase recruitment domain-containing protein 9–B cell lymphoma 10–mucosa-associated lymphoid tissue lymphoma translocation protein 1) complex34,35; in mice, this process has been shown to involve SYK-mediated phosphorylation of CARD9 by protein kinase Cδ (PKCδ; not shown)118 but is otherwise not well defined. The CARD9–BCL-10–MALT1 scaffold mediates the activation of the canonical nuclear factor-κB (NF-κB) pathway via IκB kinase subunit-β (IKKβ)-mediated degradation of NF-κB inhibitor-α (IκBα), leading to the nuclear translocation of canonical p65–p50 and REL–p50 complexes30,31,34. Dectin 1 triggering also activates the non-canonical NF-κB pathway, which is dependent on SYK activation, leading to NF-κB-inducing kinase (NIK)- and IKKα-mediated phosphorylation and proteasomal processing of NF-κB subunit p100, resulting in nuclear translocation of the non-canonical RELB–p52 complexes30,31,34. Simultaneous activation of RAF1 by dectin 1 leads to phosphorylation and acetylation (a) of p65, which enhances its transcription rate and its stability. Furthermore, phosphorylated p65 interacts with RELB, forming transcriptionally inactive RELB–p65 dimers. As RELB–p52 complexes interfere with transcription initiation of IL1B and IL12B (encoding interleukin-1β (IL-1β) and the IL-12 subunit p40 (IL-12p40), respectively), this allows for p65–p50 and REL–p50 to induce expression of IL-12p40 and pro-IL-1β, as well as IL-6, IL-12p35 and IL-23p19 (Ref. 30). SYK-dependent signalling also induces nuclear translocation of interferon-regulatory factor 1 (IRF1), which is crucial for nucleosome remodelling at the IL12A (encoding IL-12p35) promoter to enable p65–p50 binding for transcription activation49. SYK-mediated activation of TEC kinase is involved in the formation and activation of a non-canonical caspase 8 inflammasome containing the adaptor protein ASC, CARD9, BCL-10, MALT1 and caspase 8, which mediates the processing of pro-IL-1β into bioactive IL-1β44,46. The cooperation between SYK- and RAF1-mediated signalling pathways leads to expression of a cytokine profile that promotes antifungal T helper 1 (TH1) and TH17 cell differentiation. SYK-dependent signalling also induces activation of IRF5, which promotes the expression of interferon-β (IFNβ)38,39; autocrine type I IFN receptor (IFNAR) signalling then attenuates expression of TH17-polarizing IL-1β while enhancing expression of TH17-suppressing IL-27p28 via transcriptional modulation of IL27A39. This signalling pathway dampens TH17 cell differentiation40, possibly to prevent pathogenic hyperinflammation caused by TH17 cells. NEMO, NF-κB essential modulator. Dotted lines denote indirect effects.
In human DCs, p65–p50 and REL–p50 complexes trigger expression of the genes encoding IL-6 and the IL-23 subunit p19 (IL-23p19), which are necessary for TH17 cell-mediated immune responses30. Expression of IL-1β and the IL-12p40 subunit (encoded by IL12B) is equally essential to TH17 cell induction, but requires dectin 1-mediated activation of a second, SYK-independent pathway through the serine/threonine kinase RAF1 in human DCs30. RAF1 activation overcomes the repressive effects of RELB–p52 complexes on IL1B and IL12B transcription by inducing the phosphorylation and acetylation of SYK-induced p65 at Ser276 (Ref. 37), which leads to the formation of an inactive RELB–p65 dimer that is unable to bind the DNA and allows transcriptional activation of IL1B and IL12B by REL–p50 and p65–p50, respectively, in human DCs30. Furthermore, acetylated p65 has an enhanced transcription rate and is protected from degradation, further increasing transcription of the pro-inflammatory genes37 (Fig. 2). Cooperation between the distinct SYK- and RAF1-dependent signalling pathways induced via fungal recognition by dectin 1 results in a cytokine environment that directs TH17 cell differentiation30.
As aberrant TH17 cell responses are associated with pathogenic hyperinflammatory disorders9, it is important that TH17 cell differentiation is tightly controlled. One way to achieve this is through tight regulation of IL-1β production. Transcription of IL1B is regulated by controlling the activation of different NF-κB complexes (as described above), but also through autocrine type I IFN receptor (IFNAR) signalling (Fig. 2). The recognition of Candida albicans β-glucans by dectin 1 triggers SYK-dependent interferon-regulatory factor 5 (IRF5) activation and the subsequent expression of interferon-β (IFNβ)38, which results in the downregulation of IL-1β expression and the induction of IL-27 that together downregulate TH17 cell responses39,40.
A second means of controlling TH17 cell differentiation is to regulate the generation of mature IL-1β, as immature pro-IL-1β protein requires enzymatic processing to generate biologically active IL-1β for secretion41. Many pathogens, including fungi, activate cytosolic sensors, such as NLRP3 (NOD-, LRR- and pyrin domain-containing 3), to trigger the activation of canonical caspase 1 inflammasomes that cleave pro-IL-1β42. Several studies have shown that some fungi trigger the canonical caspase 1 inflammasome via dectin 1 and that this induction is dependent on fungal internalization, the production of radical oxygen species and potassium efflux43,44,45. Interestingly, dectin 1-mediated signalling also controls the processing of pro-IL-1β through activation of the non-canonical caspase 8 inflammasome44,46,47,48 (Fig. 2). The caspase 8 inflammasome is directly activated by the dectin 1 signalling pathway in response to β-glucans, independently of pathogen internalization44. Thus, dectin 1 triggering immediately couples the sensing of pathogens with activation of the non-canonical caspase 8 inflammasome, whereas some fungi also trigger the caspase 1 inflammasome via dectin 1-mediated uptake; however, it is unclear why only certain fungi trigger the canonical inflammasome.
Cooperative SYK and RAF1 signalling following dectin 1 triggering also leads to antifungal TH1 cell responses that are dependent on IL-12p70 (Ref. 30) (Fig. 2). Transcription of IL12A, encoding the IL-12p35 subunit, depends on chromatin remodelling to allow the binding of transcriptional activators such as NF-κB39. Fungal recognition by dectin 1 activates IRF1 via SYK-dependent signalling, and this promotes nucleosome remodelling at the IL12A promoter49. Moreover, the transcription of IL12A and IL12B is prolonged by RAF1-induced phosphorylation and acetylation of p65 (Ref. 30) (Fig. 2). Inhibition of RAF1 signalling30 or of IRF1 activation49 shifts dectin 1-triggered TH1 cell responses towards TH2 cell responses, which are detrimental for antifungal immunity50. Furthermore, in human neonatal DCs, dectin 1 signalling via SYK and RAF1 induces TH1 cell responses by facilitating IL12A expression51; in mouse DCs, dectin 1 signalling leads to reduced expression of OX40L, thereby shifting the response away from TH2 cell differentiation52.
RAF1 activation by dectin 1 also induces epigenetic reprogramming in monocytes, thereby enhancing innate antifungal immunity by monocytes upon a second exposure to the pathogen53, and it is conceivable that this process is important in reinforcing adaptive immunity. Collectively, these observations show that the cooperation between signalling pathways induced by dectin 1 is essential for shaping antifungal TH cell responses.
Glycan-specific TH cell responses by DC-SIGN
DC-SIGN (DC-specific ICAM3-grabbing non-integrin; also known as CD209) is expressed by subepithelial DC subsets, including CD1c+ DCs and inflammatory DCs, as well as certain macrophage subsets, and is the most extensively studied CLR17 (Table 1). Although activation of DC-SIGN by carbohydrates does not induce cytokine expression, it modulates signalling pathways that are induced by other PRRs. Many intracellular pathogens, such as mycobacteria, viruses and fungi, bind DC-SIGN via mannose-containing structures, and DC-SIGN also recognizes extracellular pathogens, such as parasites and Helicobacter pylori, via fucose-containing pathogen-associated molecular patterns (PAMPs)17. In addition to triggering DC-SIGN, these pathogens simultaneously trigger other PRRs, including TLRs and/or cytosolic receptors, and it is the cooperation between these signalling pathways that promotes protective TH1 and TH17 cell responses to intracellular pathogens or TH2 and TFH cell responses to extracellular pathogens (Fig. 3).

In immature dendritic cells (DCs), the cytoplasmic tail of DC-SIGN (DC-specific ICAM3-grabbing non-integrin) is bound by the adaptor protein LSP1 and the so-called RAF1 signalosome, consisting of CNK1, KSR1 and RAF1 (Ref. 54). a | Following recognition of mannose- containing pathogens (such as Candida albicans or mycobacteria) by DC-SIGN, various proteins are recruited to the signalosome that regulate the activation of RAF1. GTPase leukaemia-associated RHO guanine nucleotide exchange factor (LARG) activates the GTPases RAS and RHOA37,54,111. Binding of GTP–RAS to RAF1 initiates RAF1 activation by releasing intramolecular autoinhibition. Next, GTP–RHOA activates PAK1, which phosphorylates (P) RAF1 at Ser338. RAF1 is fully activated by Tyr340/341 phosphorylation through an unidentified SRC kinase54. The pathway downstream of RAF1 has yet to be fully delineated (indicated by a dashed arrow), but results in the phosphorylation at Ser276 and acetylation (a) of nuclear factor-κB (NF-κB) subunit p65 (Ref. 37). p65–p50 complexes are activated by simultaneous ligation of Toll-like receptors (TLRs). Acetylated p65 has an enhanced transcription rate and is more stable, which increases the expression of its target genes, promoting T helper 1 (TH1) and TH17 cell differentiation. b | Following recognition of fucose-containing pathogens, such as parasites and Helicobacter pylori, the RAF1 signalosome is displaced. LSP1 remains bound to DC-SIGN and becomes phosphorylated on Ser252 by MK2, which is activated by a simultaneously triggered pattern-recognition receptor (PRR). Phosphorylated LSP1 enables the assembly of a fucose-specific signalosome, consisting of IκB kinase subunit-ɛ (IKKɛ) and the de-ubiquitinase CYLD. CYLD continuously de-ubiquitylates B cell lymphoma 3 (BCL-3), preventing its nuclear accumulation. IKKɛ activation via fucose signalling leads to phosphorylation and deactivation of CYLD, which allows nuclear translocation of ubiquitylated BCL-3. Nuclear BCL-3 can positively and negatively regulate NF-κB complexes, and overall this pathway results in the modulation of TLR-induced cytokine and chemokine expression profiles to promote TH2 cell responses57. IKKɛ also phosphorylates the transcription factor signal transducer and activator of transcription 1 (STAT1) on Ser708 and Ser727 (Ref. 11). Following the induction of type I interferon receptor (IFNAR) signalling by TLR-induced interferon-β (IFNβ), JAK1-mediated phosphorylation of STAT1 at Tyr701 leads to STAT1–STAT2 dimerization, the formation of the IFN-stimulated gene factor 3 (ISGF3) complex with IFN-regulatory factor 9 (IRF9) and its translocation into the nucleus. This complex enhances the expression of interleukin-27 p28 subunit (IL-27p28), which is crucial for IL-27-mediated T follicular helper cell (TFH cell) differentiation11. CCL, CC-chemokine ligand; LPS, lipopolysaccharide.
DC-SIGN–mannose interactions direct TH1 and TH17 cell responses. DC-SIGN recognizes a diverse array of mannose PAMPs: Mycobacterium tuberculosis and Mycobacterium leprae express mannose-capped lipoarabinomannan (ManLAM) structures; many different viruses, including HIV-1, Ebola virus and hepatitis C virus, have envelope glycoproteins that contain high-mannose structures; and the cell wall of many different fungi contains the polycarbohydrate mannan17 (Table 1). These PAMPs also trigger PRRs such as TLR2, TLR4 and dectin 1, thereby activating p65–p50 transcriptional complexes and, consequently, a cytokine expression programme that results in the transcription of pro-inflammatory cytokines such as IL-12p35, IL-12p40 and IL-6 (Refs 30,37,54). DC-SIGN constitutively binds to RAF1 as part of the so-called RAF1 signalosome. Simultaneous activation of DC-SIGN and other PRRs results in the activation of RAF1 (Refs 30,54,55) and the subsequent phosphorylation and acetylation of NF-κB subunit p65 at Ser276 (Ref. 37); this enhances TLR-induced transcription of IL12A, IL12B and IL6 (Ref. 37), and the resultant increased production of active IL-12p70 promotes TH1 cell responses54 (Fig. 3a). Although IL1B is not directly induced by p65, the phosphorylation of p65 at Ser276 by RAF1 (triggered by DC-SIGN and also by dectin 1 in response to C. albicans) sequesters RELB into inactive RELB–p65 dimers, thus preventing RELB–p52-mediated inhibition of IL1B transcription30. This is important in fungal infections for which the activation of RAF1 by both dectin 1 and DC-SIGN promotes both TH1 and TH17 cell responses through the upregulation of IL-6, IL-1β and IL-23 (Refs 30,54) (Fig. 3a). Little is known about the signalling by mouse homologues of DC-SIGN, but it has been shown that in mice infected with M. tuberculosis, triggering of the DC-SIGN homologue SIGNR3 (also known as CD209D) similarly activates RAF1, and SIGNR3 signalling cooperates with TLR2 signalling to induce pro-inflammatory cytokine expression and protective immunity56. Thus, mannose signalling by DC-SIGN activates a pro-inflammatory cytokine programme via RAF1 activation.
DC-SIGN–fucose interactions direct TH2 and TFH cell responses. In contrast with mannose PAMPs, the recognition of fucose PAMPs by DC-SIGN elicits adaptive TH2 and TFH cell responses, resulting in humoral immunity11,57. These differences in adaptive immune programming probably reflect differences in mannose and fucose binding to the carbohydrate recognition domain of DC-SIGN58. Fucose binding induces conformational changes that lead to the displacement of the RAF1 signalosome from DC-SIGN, enabling the assembly of a different signalling complex termed the fucose signalosome57 (Fig. 3b). DC-SIGN recognizes a wide range of fucose PAMPs, including Lewis X antigen (Lex) from Schistosoma mansoni, fucosylated N,N′-diacetyllactosamine (LDNF) antigens from Fasciola hepatica and Ley from H. pylori17,57. H. pylori can switch on and off the expression of Lewis antigens on lipopolysaccharide (LPS), and it therefore expresses LPS with or without Ley; interestingly, Ley+ LPS, in contrast with Ley− LPS, induces TH2 cell responses via DC-SIGN signalling59 (Table 1).
Assembly of the fucose signalosome, which consists of the serine/threonine kinase IκB kinase subunit-ɛ (IKKɛ) and the de-ubiquitinase CYLD, leads to the activation and nuclear translocation of the atypical NF-κB family member, BCL-3 (Ref. 57) (Fig. 3b). BCL-3 is unable to modulate transcription by itself; however it forms a complex with p50–p50 dimers, which have a high affinity for DNA, and replaces TLR- or RIG-I-like receptor (RLR)-induced p65–p50 transcriptional complexes from promoter regions60. The effects of BCL-3–p50–p50 complexes on transcription depend on the context of the entire promoter61,62. Binding of BCL-3–p50–p50 to either the IL12A or IL12B promoter represses transcription and thus represses IL-12p70 expression, which skews TH cell responses away from a TH1 and towards a TH2 cell profile57. TH2 cell responses are further enhanced by the BCL-3–p50–p50-mediated expression of two TH2 cell-attracting chemokines, CC-chemokine ligand 17 (CCL17) and CCL22 (Ref. 57). Indeed, specific fucose PAMPs from S. mansoni, F. hepatica and H. pylori have been shown to induce TH2 cell responses in a BCL-3-dependent manner57. S. mansoni-derived PAMPs also induce OX40L expression (a marker for DCs primed for TH2 cell differentiation), possibly via DC-SIGN63.
Fucose-specific DC-SIGN signalling also leads to the expression of IL-27 (Ref. 11), which promotes TFH cell differentiation (Fig. 3b). Notably, the expression of this cytokine is independent of BCL-3 activation, but depends on crosstalk with IFNAR signalling triggered by IFNβ induced via other PRRs11. Furthermore, IKKɛ in the fucose signalosome cooperates with IFNAR signalling to induce formation of the IFN-stimulated gene factor 3 (ISGF3) transcriptional complex11, which positively regulates the expression of IL-27. Simultaneous induction of both TH2 and TFH cell responses by parasitic fucose-containing PAMPs via DC-SIGN leads to robust protective long-term humoral immunity. When TH cells were induced to differentiate in vitro by human DCs primed with fucose PAMPs, these TH cells provided cognate help to B cells and induced class switching, the production of IgG and prolonged survival11.
Moreover, fucose-specific signalling might also be important in immune tolerance, as DC-SIGN+ macrophages64 strongly upregulate IL-10 expression in response to fucose ligands, resulting in the induction of regulatory T cells (Table 1). The induction of immunity or tolerance in response to fucose-specific PAMPs might be dependent on the DC or macrophage subset, or on the co-ligation of different PRRs.
Dectin 2 promotes TH17 and TH2 cell responses
Unlike dectin 1 and DC-SIGN, dectin 2 is unable to signal by itself, instead associating with the signalling adaptor Fc receptor common γ-chain (FcRγ), which induces signalling via its ITAM65. Dectin 2 is expressed by various myeloid cell populations, including macrophages and several DC subsets, where it interacts with α-1,2-mannose structures on fungi66 and with mycobacterial ManLAM67 (Table 1).
In mice, triggering of dectin 2 on DCs by α-mannoses from C. albicans hyphae leads to SYK- and CARD9–BCLl-10–MALT1-dependent NF-κB activation68,69. Mouse dectin 2 activates p65-containing NF-κB complexes (although other subunits were not directly studied69) and induces the expression of cytokines, such as IL-6, IL-23, IL-12 and IL-1β68,69,70, resulting in the induction of TH17 cell responses69,70 (Fig. 4a). Furthermore, dectin 2–FcRγ complexes are thought to form functional complexes with the related CLR macrophage C-type lectin (MCL) in mice71. Mouse dectin 2 also induces the expression of pro-inflammatory cytokines, such as tumour necrosis factor (TNF), IL-6 and IL-23, as well as anti-inflammatory cytokines IL-10 and IL-2, in response to mycobacterial ManLAM, thereby promoting TH17 cell responses67. Interestingly, recognition of S. mansoni PAMPs by mouse dectin 2 fails to induce Il1b transcription; however, it does result in the SYK-dependent production of reactive oxygen species and potassium efflux, which activate the NLRP3–caspase 1 inflammasome to process immature pro-IL-1β produced in response to triggering of other PRRs, thereby enhancing TH17 cell responses72. Furthermore, dectin 2 signalling in mouse cells indirectly enhances dectin 1-dependent TH1 cell responses in response to fungal PAMPs, but it remains to be determined which cytokines are involved in this effect69,70. Mouse dectin 2 has also been shown to interact with glycans on allergens from Aspergillus fumigatus and the house dust mites Dermatophagoides farinae and Dermatophagoides pteronyssinus, thereby inducing signals that skew TH cell differentiation towards TH2 cell responses73,74,75 (Table 1). Recognition of D. farinae allergens by dectin 2 leads to SYK-dependent cysteinyl leukotriene production74,75, which can counteract the induction of IL-12 expression76, thereby inducing TH2 cell responses74 (Fig. 4a). Notably, sensitization of dectin 2-deficient mice with D. farinae allergens did not induce eosinophilic and neutrophilic pulmonary inflammation or TH2 cell responses in lymph nodes and lungs73. Thus, dectin 2-mediated control of TH cell differentiation in mice varies with pathogen species and ligand, and this variation might be harnessed to direct immunological responses during vaccine design.

Dectin 2 requires an association with the Fc receptor common γ-chain (FcRγ), a membrane-associated adaptor protein that contains traditional immunoreceptor tyrosine-based activation motifs (ITAMs), to induce intracellular signalling. a | Ligand recognition by mouse dectin 2 results in phosphorylation (P) of FcRγ ITAMs, which bind SHP2. Phosphorylation of the ITAMs by SHP2 facilitates the binding and activation of SYK and assembly of the CARD9–BCL-10–MALT1 (caspase recruitment domain- containing protein 9–B cell lymphoma 10–mucosa-associated lymphoid tissue lymphoma translocation protein 1) scaffold. Downstream signalling leads to the activation of nuclear factor-κB (NF-κB) p65-containing complexes, which in turn results in the expression of the T helper 17 (TH17)-polarizing cytokines pro-interleukin-1β (pro-IL-1β), IL-6 and IL-23 (Refs 67,68,69,70). By contrast, in a SYK-dependent manner, dectin 2 recognition of allergens leads to the generation of cysteinyl leukotrienes (Cys-LTs), which skews TH cell differentiation towards pathogenic TH2 cell responses73,74,75, possibly by blocking the expression of IL-12 induced by other pattern-recognition receptors (PRRs) such as Toll-like receptors (TLRs)76. b | In humans, activation of dectin 2 by fungi leads to FcRγ signalling via SHP2–SYK and the CARD9–BCL-10–MALT1 scaffold36,77. Engagement of dectin 2 alone leads to REL–p50 activation, which enhances the expression of pro-IL-1β and the IL-23 p19 subunit (IL23p19)77. Human dectin 2 can therefore only enhance TH17 cell responses that have been induced by another PRR such as dectin 1. A. fumigatus, Aspergillus fumigatus; C. albicans, Candida albicans; D. farinae, Dermatophagoides farinae; D. pteronyssinus, Dermatophagoides pteronyssinus; IκBα, NF-κB inhibitor-α; LPS, lipopolysaccharide.
Recognition of fungal pathogens by human dectin 2 also triggers signalling via FcRγ: like dectin 1 and mouse dectin 2, human dectin 2 induces a SYK-dependent CARD9–BCL-10–MALT1 scaffolding formation; however, unlike mouse dectin 2, human dectin 2 activates only REL–p50 transcriptional complexes77 (Fig. 4b). Therefore, human dectin 2 is not able to induce TH cell responses by itself, as it only induces the expression of REL-controlled interleukins, such as IL-23p19 and IL-1β, and thereby can only enhance TH17 cell responses that have been induced by other PRRs, such as dectin 1 (Refs 69,70,77,78). Human dectin 2 has also been shown to induce pro-inflammatory cytokines such as IL-1β and TNF in DCs in response to Mycobacterium bovis bacille Calmette–Guérin (BCG), but whether dectin 2 could induce these cytokines on its own was not investigated, and how this affects TH cell differentiation was not determined67. The observed differences between mouse and human dectin 2 signalling might reflect differences in species, DC subsets (bone marrow-derived DCs versus monocyte-derived DCs) or cooperation with other CLRs such as MCL.
MINCLE modulates TH1 and TH17 cell responses
Macrophage-inducible C-type lectin receptor (MINCLE; also known as CLEC4E) was first identified as a sensor of cell death79, and a recent study showed that by sensing necroptosis, MINCLE suppresses antitumor immunity and thereby enables pancreatic ductal adenocarcinoma in mice80. However, it is now evident that MINCLE, which is expressed by various immune cells (including DCs, macrophages and neutrophils) is also involved in the induction of immunity to pathogens, such as fungi and M. tuberculosis49,78,81,82. MINCLE has specificity for α-mannosyl structures and recognizes Malassezia spp. through glycolipids that contain α-mannosyl and glucosyl structures66, whereas a specific mycobacterial glycolipid known as mycobacterial cord factor (also known as TDM) is bound via its trehalose structure83 (Table 1). Interestingly, the function of MINCLE seems to be partially dependent on MCL, as this CLR controls the expression of MINCLE through the formation of heterocomplexes that stabilize MINCLE at the surface84,85. A recent study suggests that MINCLE can act independently of MCL49, but its function can be modulated by MCL co-expression86. MCL-mediated stabilization increases expression of MINCLE and thereby influences its signalling to different pathogens. Expression of MINCLE on mouse immature DCs is low, but it is upregulated by various stimuli, including the activation of MCL, whereas human immature DCs express high levels of both MINCLE and MCL49,86, suggesting that cells expressing low levels of MINCLE might be more dependent on MCL co-ligation.
Pathogen recognition by MINCLE can lead to a variety of immune responses, and it is not yet clear what underlies these differences. Like dectin 2, MINCLE associates with FcRγ for signalling79, but triggering of this CLR can result in the activation of different signalling pathways, which seem to depend partly on the ligand or DC subset involved. In mice, the recognition of mycobacterial cord factor by MINCLE together with MCL induces signalling, via FcRγ, that results in SYK-dependent CARD9–BCL-10–MALT1 scaffold formation, subsequent NF-κB activation and the expression of cytokines that lead to protective TH1 cell-mediated immunity82,87 (Fig. 5a). By contrast, in human DCs, MINCLE–FcRγ signalling induced by Fonsecaea spp. or mycobacterial cord factor results in SYK-dependent CARD9–BCL-10–MALT1 scaffold formation, but not NF-κB activation nor cytokine gene expression49. Instead, MINCLE triggering leads to phosphoinositide 3-kinase (PI3K)- and AKT-dependent activation of the E3 ubiquitin ligase MDM2 (Ref. 49); the action of this enzyme targets nuclear IRF1 for proteasomal degradation, thereby suppressing the production of IL-12p70 and skewing TH cell differentiation towards TH2 cell responses49 (Fig. 5b), which have adverse effects on defence against fungal infections50. Indeed, a high fungal load in lesions from patients with chromoblastomycosis is characterized by the presence of TH2 cells88. Thus, Fonsecaea spp. escape TH1 cell-mediated immunity via MINCLE. Interestingly, in a mouse model of chromoblastomycosis, co-stimulation of TLRs, such as TLR2, TLR4 and TLR7, leads to strong TNF responses and promotes an efficient MINCLE-dependent protective immunity against Fonsecaea pedroso i89. These data suggest that Fonsecaea spp. target MINCLE to suppress antifungal responses, which can be altered by co-ligation with TLRs.

Macrophage-inducible C-type lectin receptor (MINCLE) associates with the Fc receptor common γ-chain (FcRγ) to induce intracellular signalling79. a | In mice, the expression of MINCLE is upregulated on activated myeloid cells, and it associates with macrophage C-type lectin (MCL) after initial binding of mycobacteria, mycobacterial cord factor or its synthetic analogue, TDB (not shown)84,85,86. MCL also associates with FcRγ84. It remains undefined how proximal MINCLE –MCL signalling proceeds, but it is likely that SHP2 and SYK are consecutively recruited to the immunoreceptor tyrosine-based activation motifs (ITAMs) of the associated FcRγ. MINCLE –MCL recognition of mycobacterial cord factor results in the SYK-dependent formation of the CARD9–BCL-10–MALT1 (caspase recruitment domain-containing protein 9–B cell lymphoma 10–mucosa-associated lymphoid tissue lymphoma translocation protein 1) scaffold, which activates nuclear factor-κB (NF-κB) transcriptional complexes (the precise composition of which has not been described) and thus induces expression of cytokines that promote T helper 1 (TH1) and TH17 cell responses82,87,90,91. b | It is not yet clear whether MCL is involved in signalling by human MINCLE. In DCs, both MINCLE and MCL are highly expressed, suggesting that MCL might be required less for the induction of MINCLE expression in humans than in mice49,86. In humans, MINCLE has been shown to operate independently of MCL and to signal through FcRγ. SYK activation on recognition of pathogenic Fonsecaea spp. leads to CARD9–BCL-10–MALT1 scaffold formation, but not NF-κB activation49. Instead, a signalling cascade via phosphoinositide 3-kinase (PI3K) is initiated; however, how the CARD9–BCL-10–MALT1 scaffold is linked to PI3K activation remains elusive49. PI3K activation leads to activation of AKT via phosphorylation (P) of Thr308 and Ser473. AKT in the cytoplasm directly phosphorylates the E3 ubiquitin ligase MDM2 at Ser166, which promotes MDM2 translocation to the nucleus49,119,120. Within the nucleus, MDM2 associates with dectin 1-induced interferon-regulatory factor 1 (IRF1), which activates the ubiquitin ligase activity of MDM2 and thereby targets IRF1 for proteasomal degradation. This completely blocks the transcriptional activation of IL12A (encoding interleukin-12 subunit p35 (IL-12p35)) via dectin 1 or Toll-like receptor 4 (TLR4), hence skewing TH cell differentiation towards pro-fungal TH2 cell responses49. IκBα, NF-κB inhibitor-α; LPS, lipopolysaccharide.
MINCLE also affects TH17 cell-mediated immunity, seemingly in a pathogen-dependent manner, but host species differences might also be involved. Mouse MINCLE induces TH17 cell-mediated immunity to mycobacterial cord factor by enhancing expression of the TH17 cell-promoting factors IL-6, IL-23 and pro-IL-1β82,87,90 (Fig. 5a), and by activation of the NLRP3 inflammasome, thereby converting pro-IL-1β to bioactive IL-1β91. However, triggering of MINCLE in mice during F. pedrosoi infection in vivo suppresses dectin 2-mediated TH17 cell differentiation, although the underlying mechanism remains elusive78. By contrast, human MINCLE-mediated signalling seems to promote TH17 cell responses by attenuating type I IFN responses. MINCLE -induced activation of MDM2 also leads to the degradation of IRF5, which is the transcriptional activator of IFNB39. As described above, IFNβ affects both IL-1β and IL-27 expression, and hence reduced IFNβ levels increase the expression of TH17 cell-polarizing IL-1β and reduce the expression of TH17 cell-dampening IL-27, with a net effect of increased TH17 cell-mediated immunity39.
Recently, it was shown that human MINCLE, but not mouse MINCLE nor other CLRs such as MCL, recognizes cholesterol crystals and induces innate immune responses92. It has been suggested that another cholesterol sensor (or sensors) might also be involved, as not all innate immune responses were dependent on MINCLE92. Furthermore, co-ligation of LPS with cholesterol crystals enhances IL-1β expression92, suggesting that MINCLE can also cooperate with TLRs, and this cooperation with other PRRs might account for the observed differences in immune responses between host species and between infecting pathogens.
These data suggest that MINCLE has diverse roles in inducing immunity to pathogens as well as endogenous compounds. Pathogenic fungi such as Fonsecaea spp. seem to target MINCLE to modulate or suppress immunity induced by other CLRs39,78. An important factor in MINCLE function is its formation of a complex with MCL, thus stabilizing MINCLE expression at the cell surface; this might lead to the specific induction of NF-κB and subsequent pro-inflammatory cytokine responses84,85. Further research is required to delineate the signalling pathways induced by MINCLE and how MCL or other PRRs can affect its function in inducing or suppressing immunity.
Diversification with common signalling scaffolds
Although many CLRs that function as PRRs signal via SYK-mediated CARD9–BCL-10–MALT1 scaffolds, the activation of different CLRs often has distinct immunological outcomes, as discussed above. One intriguing question is how these receptors induce distinct signalling pathways through a common signalling scaffold.
Dectin 1 signalling differs vastly from FcRγ-mediated dectin 2 and MINCLE signalling, as it also triggers LSP1–RAF1-dependent signalling (S.I.G. and T.B.H.G., unpublished observations). Although research to date has mainly focused on RAF1 activation and its effect on NF-κB activation, LSP1 (either dependently or independently of RAF1) could have various effects on other signalling pathways downstream of SYK and CARD9–BCL-10–MALT1, which would distinguish dectin 1 signalling from dectin 2–FcRγ and MINCLE–FcRγ signalling. For example, human dectin 1 mediates the recruitment of TNF receptor-associated factor 2 (TRAF2) and TRAF6 to the CARD9–BCL-10–MALT1 scaffold, whereas human dectin 2 recruits only TRAF2 (S.I.G. and T.B.H.G., unpublished observations). This difference might be associated with LSP1-mediated signalling downstream of dectin 1, and may explain why triggering of dectin 1 leads to the activation of both classical p65–p50 and REL–p50 NF-κB complexes, whereas dectin 2 activates only REL–p50 complexes. However, further research is needed to confirm this hypothesis.
Differences in the complexes formed by different CLRs might also influence downstream signalling. Whereas dectin 1 is thought to form dimers35, dectin 2 and MINCLE are coupled to FcRγ and are also capable of forming functional complexes with other receptors, such as MCL, which might affect their ability to form proximal signalling complexes. For example, MINCLE–MCL complexes were shown to induce NF-κB activation via the CARD9–BCL-10–MALT1 complex, whereas MINCLE alone induces the CARD9–BCL-10–MALT1 scaffold, but does not activate NF-κB; the conformation of the scaffold in this case might be inefficient in activating the TRAF– TAK (TGFβ-activated kinase) complexes necessary for classical NF-κB activation, but sufficient to activate PI3K. Similarly, dimerization of dectin 2 with MCL71 might allow FcRγ dimerization and, subsequently, efficient CARD9–BCL-10–MALT1 scaffold formation and downstream NF-κB activation. Deeper insights into the molecular regulation of the CARD9–BCL-10–MALT1 scaffold and downstream signalling complexes will hopefully provide clarification to these intriguing aspects of CLR functions.
Other CLRs in TH cell differentiation
Several other CLRs have also been shown to induce or inhibit adaptive immunity, but the underlying mechanisms are unknown. For example, the human mannose receptor contributes to antifungal TH17 cell-mediated immunity93, whereas the recognition of different allergens by this mannose receptor results in the downregulation of TLR-induced IL-12p70 expression, thereby favouring TH2 cell responses94. MGL (also known as CLEC10A) engagement enhances TLR-induced IL-10 expression, resulting in the induction of regulatory T cells95,96. CLEC9A in mice is involved in the induction of antigen-specific TFH cells97; however, this receptor seems to be more important for antigen cross-presentation than for the production of pro-inflammatory cytokines98,99, suggesting that cooperation between CLEC9A and other receptors might underlie the observed effects on TFH cell responses. Over the next few years, it will become clear whether the engagement of specific CLRs is also involved in the induction of other specialized TH cell subsets, such as TH9 and TH22 cells.
Harnessing CLRs for vaccine design
There is an urgent need for vaccines that protect against infections such as HIV-1 and malaria or against emerging threats such as Ebola virus, as well as against cancer. Although most successful vaccines confer protection through neutralizing antibodies, it is becoming clear that cell-mediated immunity is equally important. CLRs have long been studied as vaccine targets to improve antigen presentation100. Targeting of the CLR DEC205 (also known as LY75) via an antibody linked to an antigen induces antigen-specific CD4+ and CD8+ T cells101,102,103. The presence or absence of adjuvants (such as TLR ligands or CD40L) determines whether these antigen-specific responses induce immunity or tolerance, respectively101,102. In these studies, the main focus has been on the antigen presentation ability of CLRs and not their ability to induce immunity. However, there is a large diversity in the possible immune responses induced by CLRs, which depend on the type of CLR and DC subset, the nature of the carbohydrate ligand, and importantly, the co-ligation with another receptor (or receptors), such as another PRR. This diversity makes CLRs ideal vaccine targets for immune modulation.
There are several possible means of triggering CLRs and inducing specific immunity, such as attenuated or dead pathogens, carbohydrate ligands or particles containing both carbohydrate ligands and adjuvants (Fig. 6). As discussed above, the recognition of pathogens by CLRs leads to specific immune responses (Table 1). However, because pathogens trigger different CLRs on distinct DC and macrophage subsets, it is difficult to predict the type of immune response that will be elicited. Although whole pathogens that trigger CLRs, such as M. bovis BCG and live attenuated viruses, have been used successfully in the design of vaccines104, the wide variety of possible CLR–PRR crosstalk in such an approach predicts that this strategy will probably not be successful for more complex pathogens or tumours.

C-type lectin receptor (CLR) targeting not only enhances antigen presentation but also may offer an approach to target specific T helper cell (TH cell)-mediated responses to vaccines against a pathogen or disease of choice. Different methods of targeting CLRs have been studied, including antigen-linked antibodies, glycosylated antigens, and glycosylated particles containing both antigens and possible adjuvants such as Toll-like receptor (TLR) ligands. PRR, pattern-recognition receptor; TCR, T cell receptor.
An alternative might be the use of single carbohydrate structures or microbial glycoproteins or glycolipids. β-glucans are good candidates, as they are specifically recognized by dectin 1 and induce protective TH1 and TH17 cell immunity to C. albicans infections25,30,31,34. Furthermore, β-glucans have been successfully used to enhance antitumor responses, confirming their potential in this capacity105. Mycobacterial cord factor appears to be a specific ligand for MINCLE–MCL and has successfully been used in mice for vaccinations against mycobacterial infections87,106. Moreover, viral envelope glycoproteins allow the targeting of several CLRs, such as DC-SIGN, langerin, DC immunoreceptor (DCIR; also known as CLEC4A) and mannose receptor, and when used in combination with TLR ligands, these glycoproteins have been shown to modulate innate signalling that shapes immunity37,54,107. Helminth parasites express a large variety of glycoproteins, glycolipids and carbohydrates that are recognized by CLRs such as DC-SIGN, mannose receptor and MGL, and that can induce specific immune responses108,109. Their interaction with CLRs can explain the beneficial use of helminth eggs in anti-inflammatory treatment of Crohn disease110.
A drawback of using single carbohydrate structures is that many CLRs have overlapping binding specificities17, which does not allow for specific CLR targeting and may potentially induce distinct and even contrasting immune responses. By contrast, antibodies against CLRs are by definition specific and therefore useful in targeting CLRs. Their use has been extensively investigated for enhancing antigen presentation, but their ability to trigger CLR signalling has been only marginally investigated. A few soluble antibodies are known to induce CLR signalling: the CLEC9A-trageting antibody 24/04-10B4 has been shown to induce TFH cell responses97, and the polyclonal DC-SIGN-targeting antibody H-200 triggers signalling similar to that of mannose ligands111. The high specificity of antibodies underscores their possible application in triggering CLRs.
Recent studies have shown that broadly neutralizing antibodies against HIV-1 can be protective against HIV-1 infection112, but it is thought that many rounds of TFH cell-mediated selection of B cells in germinal centres are necessary to form these broadly neutralizing antibodies113. Therefore, vaccines that induce efficient TFH cell responses might elicit these broadly neutralizing antibodies. Interestingly, in contrast to other PRRs, some CLRs, such as DC-SIGN and CLEC9A, have been shown to specifically elicit TFH cell responses11,97. Thus, CLR targeting might also present an important opportunity to induce strong antibody responses against HIV-1.
Tumour research has focused on targeting CLRs for enhancing antigen presentation and, in particular, cross-presentation for CD8+ T cell activation114,115, with little focus on targeting CLRs for their immunomodulatory traits. Interestingly, tumours exhibit aberrant glycosylation; for example, certain tumours express fucose-containing antigens or sialic acids that suppress immunity116,117. Thus, a vaccine aimed at counteracting the immunosuppressive tumour environment might offer an effective strategy. For instance, the induction of TH1 cell responses in combination with cross-presentation might be achieved by the use of liposomes carrying TLR ligands and/or adjuvants along with agonist antibodies or carbohydrates specific for CLRs that induce TH1 cell responses. Indeed, targeting of dectin 1 to enhance ovalbumin (OVA)-specific TH1 and TH17 cell responses has been effective against an OVA-expressing tumour in mice105. Further research will show whether both the antigen presentation capacities of CLRs and their immunomodulatory functions can be harnessed to combat tumours and infectious diseases.
Using CLR-targeted vaccines in combination with other vaccine platforms, such as nanoparticles or DNA vaccines, could further improve vaccine development. CLR targeting allows for the induction of specific adaptive immunity, so the use of CLR-targeting molecules as adjuvants can increase the effectiveness of other vaccines, such as DNA vaccines, that enhance antigen presentation. Moreover, other vaccination strategies can be modulated to target CLRs. An interesting possibility would be to modulate the glycosylation of viral-vector-based vaccines or attenuated viruses such that the glycoproteins interact with specific CLRs and/or are excluded from binding to other CLRs, and in this way to both shape adaptive immunity and enhance antigen uptake and presentation.
Thus, despite or perhaps because of the complexity of CLR signalling, CLR targeting provides a plethora of possibilities in vaccine design rationale, in a similar manner to how successful pathogens and tumours have ingeniously evolved to escape immunity.
Concluding remarks
Together, CLRs induce a breadth of TH cell responses that allows for a level of adaptive immune diversification not seen with other PRRs. Recent progress from research on CLR signalling and how this tailors efficient protective immunity to pathogens has been very important for allowing a more rational consideration of the glycoprotein- or carbohydrate-conjugated antigens and adjuvants used in developing vaccines. CLRs have been extensively studied as targets to enhance antigen presentation and the induction of antigen-specific T cells, but now the ability of CLRs to shape adaptive immunity to pathogens should be exploited in vaccination strategies.
References
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010).
Raphael, I., Nalawade, S., Eagar, T. N. & Forsthuber, T. G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 74, 5–17 (2015).
Wynn, T. A. Type 2 cytokines: mechanisms and therapeutic strategies. Nat. Rev. Immunol. 15, 271–282 (2015).
O'Shea, J. J., Lahesmaa, R., Vahedi, G., Laurence, A. & Kanno, Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat. Rev. Immunol. 11, 239–250 (2011).
Pulendran, B., Tang, H. & Manicassamy, S. Programming dendritic cells to induce TH2 and tolerogenic responses. Nat. Immunol. 11, 647–655 (2010).
Wang, Y. H. & Liu, Y. J. Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clin. Exp. Allergy 39, 798–806 (2009).
Eyerich, S. et al. IL-22 and TNF-α represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur. J. Immunol. 41, 1894–1901 (2011).
Manel, N., Unutmaz, D. & Littman, D. R. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat. Immunol. 9, 641–649 (2008).
Gaffen, S. L., Jain, R., Garg, A. V. & Cua, D. J. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600 (2014).
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Gringhuis, S. I. et al. Fucose-based PAMPs prime dendritic cells for follicular T helper cell polarization via DC-SIGN-dependent IL-27 production. Nat. Commun. 5, 5074 (2014). This paper shows how cooperation between fucose-specific DC-SIGN signalling and IFNAR signalling drives T FH cell differentiation; this paper also identifies IL-27 as an important cytokine for human T FH cell induction.
Batten, M. et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 207, 2895–2906 (2010).
Kaplan, M. H., Hufford, M. M. & Olson, M. R. The development and in vivo function of T helper 9 cells. Nat. Rev. Immunol. 15, 295–307 (2015).
Jia, L. & Wu, C. The biology and functions of Th22 cells. Adv. Exp. Med. Biol. 841, 209–230 (2014).
Shevach, E. M. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645 (2009).
Drickamer, K. & Taylor, M. E. Recent insights into structures and functions of C-type lectins in the immune system. Curr. Opin. Struct. Biol. 34, 26–34 (2015).
Geijtenbeek, T. B. & Gringhuis, S. I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009).
Taylor, P. R. et al. The β-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 169, 3876–3882 (2002).
van den Berg, L. M., Zijlstra-Willems, E. M., Richters, C. D., Ulrich, M. M. & Geijtenbeek, T. B. Dectin-1 activation induces proliferation and migration of human keratinocytes enhancing wound re-epithelialization. Cell. Immunol. 289, 49–54 (2014).
Poulin, L. F. et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 207, 1261–1271 (2010).
Valladeau, J. et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12, 71–81 (2000).
Stansell, E. & Desrosiers, R. C. Fundamental difference in the content of high-mannose carbohydrate in the HIV-1 and HIV-2 lineages. J. Virol. 84, 8998–9009 (2010).
Ju, T., Aryal, R. P., Kudelka, M. R., Wang, Y. & Cummings, R. D. The Cosmc connection to the Tn antigen in cancer. Cancer Biomark. 14, 63–81 (2014).
Ferwerda, B. et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 361, 1760–1767 (2009).
Taylor, P. R. et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat. Immunol. 8, 31–38 (2007).
Saijo, S. et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 8, 39–46 (2007).
Marakalala, M. J. et al. Differential adaptation of Candida albicans in vivo modulates immune recognition by dectin-1. PLoS Pathog. 9, e1003315 (2013).
Tang, C. et al. Inhibition of dectin-1 signaling ameliorates colitis by inducing Lactobacillus-mediated regulatory T cell expansion in the intestine. Cell Host Microbe 18, 183–197 (2015). This report demonstrates the importance of dectin 1 in regulating the homeostasis of intestinal immunity.
Kashem, S. W. et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 42, 356–366 (2015).
Gringhuis, S. I. et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-κB activation through Raf-1 and Syk. Nat. Immunol. 10, 203–213 (2009).
LeibundGut-Landmann, S. et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8, 630–638 (2007). This work determines the importance of dectin 1 signalling in the induction of T H 17 cells in vivo.
Goodridge, H. S. et al. Activation of the innate immune receptor Dectin-1 upon formation of a 'phagocytic synapse'. Nature 472, 471–475 (2011).
Gantner, B. N., Simmons, R. M., Canavera, S. J., Akira, S. & Underhill, D. M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 197, 1107–1117 (2003).
Gross, O. et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656 (2003).
Rogers, N. C. et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22, 507–517 (2005). This investigation finds that dectin 1 activates SYK and can thereby induce immunity independently of TLR signalling.
Deng, Z. et al. Tyrosine phosphatase SHP-2 mediates C-type lectin receptor-induced activation of the kinase Syk and anti-fungal TH17 responses. Nat. Immunol. 16, 642–652 (2015). This research shows that the tyrosine phosphatase SHP2 acts as a scaffold for the recruitment of SYK to dectin 1 or to the adaptor FcRγ, thereby controlling immunity induced by different CLRs.
Gringhuis, S. I. et al. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-κB. Immunity 26, 605–616 (2007).
del Fresno, C. et al. Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38, 1176–1186 (2013).
Wevers, B. A. C-type lectin signaling in dendritic cells. Molecular control of antifungal inflammation. Thesis, Univ. Amsterdam (2014).
Liu, H. & Rohowsky-Kochan, C. Interleukin-27-mediated suppression of human Th17 cells is associated with activation of STAT1 and suppressor of cytokine signaling protein 1. J. Interferon Cytokine Res. 31, 459–469 (2011).
Dinarello, C. A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 (2009).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Saïd-Sadier, N., Padilla, E., Langsley, G. & Ojcius, D. M. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE 5, e10008 (2010).
Gringhuis, S. I. et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254 (2012). This paper identifies a caspase 8-containing complex that is induced by fungal and mycobacterial infections and that leads to direct processing of IL-1 β , independently of caspase 1.
Cheng, S. C. et al. The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J. Leukoc. Biol. 90, 357–366 (2011).
Zwolanek, F. et al. The non-receptor tyrosine kinase Tec controls assembly and activity of the noncanonical caspase-8 inflammasome. PLoS Pathog. 10, e1004525 (2014).
Ganesan, S. et al. Caspase-8 modulates dectin-1 and complement receptor 3-driven IL-1β production in response to β-glucans and the fungal pathogen Candida albicans. J. Immunol. 193, 2519–2530 (2014).
Rieber, N. et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe 17, 507–514 (2015).
Wevers, B. A. et al. Fungal engagement of the C-type lectin Mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 15, 494–505 (2014).
Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 11, 275–288 (2011).
Lemoine, S. et al. Dectin-1 activation unlocks IL12A expression and reveals the TH1 potency of neonatal dendritic cells. J. Allergy Clin. Immunol. 136, 1355–1368 (2015).
Joo, H. et al. Opposing roles of dectin-1 expressed on human plasmacytoid dendritic cells and myeloid dendritic cells in Th2 polarization. J. Immunol. 195, 1723–1731 (2015).
Saeed, S. et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014).
Gringhuis, S. I., den Dunnen, J., Litjens, M., van der Vlist, M. & Geijtenbeek, T. B. H. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 10, 1081–1088 (2009).
Sarkar, R., Mitra, D. & Chakrabarti, S. HIV-1 gp120 protein downregulates Nef induced IL-6 release in immature dentritic cells through interplay of DC-SIGN. PLoS ONE 8, e59073 (2013).
Tanne, A. et al. A murine DC-SIGN homologue contributes to early host defense against Mycobacterium tuberculosis. J. Exp. Med. 206, 2205–2220 (2009).
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Mesman, A. W. & Geijtenbeek, T. B. Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKɛ- and CYLD-dependent Bcl3 activation. Nat. Commun. 5, 3898 (2014).
Guo, Y. et al. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11, 591–598 (2004).
Bergman, M. P. et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J. Exp. Med. 200, 979–990 (2004).
Ghosh, S. & Hayden, M. S. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 8, 837–848 (2008).
Mühlbauer, M., Chilton, P. M., Mitchell, T. C. & Jobin, C. Impaired Bcl3 up-regulation leads to enhanced lipopolysaccharide-induced interleukin (IL)-23p19 gene expression in IL-10−/− mice. J. Biol. Chem. 283, 14182–14189 (2008).
Wessells, J. et al. BCL-3 and NF-κB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J. Biol. Chem. 279, 49995–50003 (2004).
Kuijk, L. M. et al. Soluble helminth products suppress clinical signs in murine experimental autoimmune encephalomyelitis and differentially modulate human dendritic cell activation. Mol. Immunol. 51, 210–218 (2012).
Conde, P. et al. DC-SIGN+ macrophages control the induction of transplantation tolerance. Immunity 42, 1143–1158 (2015).
Sato, K. et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor γ chain to induce innate immune responses. J. Biol. Chem. 281, 38854–38866 (2006).
Ishikawa, T. et al. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe 13, 477–488 (2013).
Yonekawa, A. et al. Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity 41, 402–413 (2014).
Bi, L. et al. CARD9 mediates Dectin-2-induced IκBα kinase ubiquitination leading to activation of NF-κB in response to stimulation by the hyphal form of Candida albicans. J. Biol. Chem. 285, 25969–25977 (2010).
Saijo, S. et al. Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32, 681–691 (2010).
Robinson, M. J. et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 206, 2037–2051 (2009).
Zhu, L. L. et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39, 324–334 (2013).
Ritter, M. et al. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc. Natl Acad. Sci. USA 107, 20459–20464 (2010).
Parsons, M. W. et al. Dectin-2 regulates the effector phase of house dust mite-elicited pulmonary inflammation independently from its role in sensitization. J. Immunol. 192, 1361–1371 (2014).
Barrett, N. A. et al. Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J. Exp. Med. 208, 593–604 (2011). This study demonstrates that dectin 2 contributes to allergic T H 2 cell responses by inducing the production of cysteinyl leukotrienes.
Barrett, N. A., Maekawa, A., Rahman, O. M., Austen, K. F. & Kanaoka, Y. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J. Immunol. 182, 1119–1128 (2009).
Machida, I. et al. Cysteinyl leukotrienes regulate dendritic cell functions in a murine model of asthma. J. Immunol. 172, 1833–1838 (2004).
Gringhuis, S. I. et al. Selective c-Rel activation via Malt1 controls anti-fungal TH-17 immunity by dectin-1 and dectin-2. PLoS Pathog. 7, e1001259 (2011).
Wuthrich, M. et al. Fonsecaea pedrosoi-induced Th17-cell differentiation in mice is fostered by Dectin-2 and suppressed by Mincle recognition. Eur. J. Immunol. 45, 2542–2552 (2015).
Yamasaki, S. et al. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188 (2008). This work identifies the ligand for MINCLE and shows the importance of this receptor as a sensor of damaged cells.
Seifert, L. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016).
Yamasaki, S. et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl Acad. Sci. USA 106, 1897–1902 (2009).
Schoenen, H. et al. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 184, 2756–2760 (2010).
Jégouzo, S. A. et al. Defining the conformation of human mincle that interacts with mycobacterial trehalose dimycolate. Glycobiology 24, 1291–1300 (2014).
Miyake, Y., Masatsugu, O. H. & Yamasaki, S. C-type lectin receptor MCL facilitates Mincle expression and signaling through complex formation. J. Immunol. 194, 5366–5374 (2015).
Lobato-Pascual, A., Saether, P. C., Fossum, S., Dissen, E. & Daws, M. R. Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FcɛRI-γ. Eur. J. Immunol. 43, 3167–3174 (2013).
Ostrop, J. et al. Contribution of MINCLE–SYK signaling to activation of primary human APCs by mycobacterial cord factor and the novel adjuvant TDB. J. Immunol. 195, 2417–2428 (2015).
Werninghaus, K. et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRγ–Syk–Card9- dependent innate immune activation. J. Exp. Med. 206, 89–97 (2009). This report describes an important role for FcR γ signalling in the induction of immune responses to mycobacterial cord factor.
d'Avila, S. C., Pagliari, C. & Duarte, M. I. The cell-mediated immune reaction in the cutaneous lesion of chromoblastomycosis and their correlation with different clinical forms of the disease. Mycopathologia 156, 51–60 (2003).
da Glória Sousa, M. et al. Restoration of pattern recognition receptor costimulation to treat chromoblastomycosis, a chronic fungal infection of the skin. Cell Host Microbe 9, 436–443 (2011).
Miyake, Y. et al. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38, 1050–1062 (2013).
Schweneker, K. et al. The mycobacterial cord factor adjuvant analogue trehalose-6,6′-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology 218, 664–673 (2013).
Kiyotake, R. et al. Human Mincle binds to cholesterol crystals and triggers innate immune responses. J. Biol. Chem. 290, 25322–25332 (2015).
van de Veerdonk, F. L. et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 5, 329–340 (2009).
Salazar, F. et al. The mannose receptor negatively modulates the Toll-like receptor 4-aryl hydrocarbon receptor-indoleamine 2,3-dioxygenase axis in dendritic cells affecting T helper cell polarization. J. Allergy Clin. Immunol. http://dx.doi.org/10.1016/j.jaci.2015.10.033 (2015).
van Vliet, S. J. et al. MGL signaling augments TLR2-mediated responses for enhanced IL-10 and TNF-α secretion. J. Leukoc. Biol. 94, 315–323 (2013).
Li, D. et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10- producing suppressive CD4+ T cells. J. Exp. Med. 209, 109–121 (2012).
Kato, Y. et al. Targeting antigen to Clec9A primes follicular Th cell memory responses capable of robust recall. J. Immunol. 195, 1006–1014 (2015).
Sancho, D. et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903 (2009).
Zelenay, S. et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Invest. 122, 1615–1627 (2012).
Steinman, R. M. Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 30, 1–22 (2012).
Bonifaz, L. et al. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638 (2002).
Hawiger, D. et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–779 (2001).
Idoyaga, J. et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl Acad. Sci. USA 108, 2384–2389 (2011).
Pulendran, B., Oh, J. Z., Nakaya, H. I., Ravindran, R. & Kazmin, D. A. Immunity to viruses: learning from successful human vaccines. Immunol. Rev. 255, 243–255 (2013).
LeibundGut-Landmann, S., Osorio, F., Brown, G. D. & Reis e Sousa, C. Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood 112, 4971–4980 (2008). This investigation shows how dectin 1 can be targeted to activate cytotoxic T cell responses.
Behler, F. et al. Macrophage-inducible C-type lectin Mincle-expressing dendritic cells contribute to control of splenic Mycobacterium bovis BCG infection in mice. Infect. Immun. 83, 184–196 (2015).
Meyer-Wentrup, F. et al. DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J. Leukoc. Biol. 85, 518–525 (2009).
van Stijn, C. M. et al. Schistosoma mansoni worm glycolipids induce an inflammatory phenotype in human dendritic cells by cooperation of TLR4 and DC-SIGN. Mol. Immunol. 47, 1544–1552 (2010).
Everts, B. et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J. Exp. Med. 209, 1753–1767, (2012).
Szkudlapski, D. et al. The emering role of helminths in treatment of the inflammatory bowel disorders. J. Physiol. Pharmacol. 65, 741–751 (2014).
Hodges, A. et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat. Immunol. 8, 569–577 (2007).
Caskey, M. et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522, 487–491 (2015).
Streeck, H., D'Souza, M. P., Littman, D. R. & Crotty, S. Harnessing CD4+ T cell responses in HIV vaccine development. Nat. Med. 19, 143–149 (2013).
Palucka, K. & Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 12, 265–277 (2012).
Fehres, C. M., Unger, W. W., Garcia-Vallejo, J. J. & van Kooyk, Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol. 5, 149 (2014).
Perdicchio, M. et al. Tumor sialylation impedes T cell mediated anti-tumor responses while promoting tumor associated-regulatory T cells. Oncotarget 7, 8771–8782 (2016).
Chen, J. T. et al. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl Acad. Sci. USA 112, 13057–13062 (2015).
Strasser, D. et al. Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 36, 32–42 (2012).
Mayo, L. D. & Donner, D. B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl Acad. Sci. USA 98, 1598–11603 (2001).
Zhou, B. P. et al. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 3, 973–982 (2001).
Chamilos, G. et al. Generation of IL-23 producing dendritic cells (DCs) by airborne fungi regulates fungal pathogenicity via the induction of TH-17 responses. PLoS ONE 5, e12955 (2010).
Toth, A. et al. Candida albicans and Candida parapsilosis induce different T-cell responses in human peripheral blood mononuclear cells. J. Infect. Dis. 208, 690–698 (2013).
Gessner, M. A. et al. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect. Immun. 80, 410–417 (2012).
Viriyakosol, S., Jimenez, M.d. P., Gurney, M. A., Ashbaugh, M. E. & Fierer, J. Dectin-1 s required for resistance to coccidioidomycosis in mice. mBio 4, e00597–00512 (2013).
Wang, H. et al. C-type lectin receptors differentially induce th17 cells and vaccine immunity to the endemic mycosis of North America. J. Immunol. 192, 1107–1119 (2014).
Loures, F. V., Araujo, E. F., Feriotti, C., Bazan, S. B. & Calich, V. L. TLR-4 cooperates with Dectin-1 and mannose receptor to expand Th17 and Tc17 cells induced by Paracoccidioides brasiliensis stimulated dendritic cells. Front. Microbiol. 6, 261 (2015).
Higashino-Kameda, M. et al. A critical role of Dectin-1 in hypersensitivity pneumonitis. Inflamm. Res. 65, 235–44 (2015).
Uryu, H. et al. α-Mannan induces Th17-mediated pulmonary graft-versus-host disease in mice. Blood 125, 3014–3023 (2015).
Norimoto, A. et al. Dectin-2 promotes house dust mite-induced T helper type 2 and type 17 cell differentiation and allergic airway inflammation in mice. Am. J. Respir. Cell. Mol. Biol. 51, 201–209 (2014).
Ishikawa, E. et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 13, 2879–2888 (2009).
Acknowledgements
S.I.G and T.B.H.G. were supported by Aids Fonds (grants 2014014, 2012042, 2010050 and 2009024), the Netherlands Organization for Scientific Research (grants NGI 40-41009-98-8057 and NWO VICI 918.10.619), the Dutch Burns Foundation (grant 08.109) and the European Research Council (Advanced grant 670424).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Germinal centres
-
Highly specialized and dynamic microenvironments that give rise to secondary B cell follicles during an immune response. They are the main sites of B cell maturation, leading to the generation of memory B cells and plasma cells that produce high-affinity antibody.
- C-type lectin receptors
-
(CLRs). Receptors that recognize carbohydrates expressed by microorganisms, as well as endogenous glycoproteins. Some CLRs function as pattern-recognition receptors, whereas others function as adhesion receptors. The recognition of microorganisms by CLRs on antigen-presenting cells leads to their internalization and subsequent antigen processing and presentation. In addition, some CLRs efficiently induce signalling pathways to promote or modulate T helper cell differentiation.
- Pattern-recognition receptors
-
(PRRs). Receptors that recognize molecular patterns often associated with pathogens, such as lipopolysaccharides found in the bacterial cell wall of Gram-negative microorganisms. They also recognize molecular patterns expressed by non-pathogenic bacteria.
- Canonical NF-κB pathway
-
The classical pathway of nuclear factor-κB (NF-κB) activation that involves phosphorylation and degradation of the NF-κB inhibitor IκBα.
- Non-canonical NF-κB pathway
-
The pathway of nuclear factor-κB (NF-κB) activation that does not involve degradation of the NF-κB inhibitor IκBα but relies on the processing of an NF-κB precursor protein, p100, leading to nuclear translocation of the RELB–p52 NF-κB heterodimer.
- Inflammasomes
-
Complexes that contain a sensor molecule, such as NLRP3 (NOD-, LRR- and pyrin domain-containing 3), the adaptor protein ASC and caspase 1. The term non-canonical inflammasome describes an inflammasome- like complex that does not conform to the three 'canonical' components but, similarly to canonical inflammasomes, processes immature pro- interleukin-1β (pro-IL-1β) to generate bio-active IL-1β for secretion. Two non-canonical inflammasomes have been described to date: one that contains mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), ASC and caspase 8 (termed the non-canonical caspase 8 inflammasome) and is triggered by dectin 1 engagement, and a caspase 11-activating platform that has not yet been fully described.
- Chromatin remodelling
-
Alterations that are induced in chromatin through the displacement of nucleosomes by ATP-dependent multiprotein complexes.
- Pathogen-associated molecular patterns
-
(PAMPs). Small conserved molecular motifs found on microorganisms, such as lipopolysaccharide (LPS), carbohydrate moieties, double-stranded RNA and unmethylated DNA motifs.
- RAF1 signalosome
-
A complex containing the serine/threonine kinase RAF1, CNK1 and KSR1. Mannose signalling activates RAF1 in this complex, triggering a downstream signalling cascade that induces T helper 1 (TH1) and TH17 cell differentiation.
- Fucose signalosome
-
A complex containing IκB kinase subunit-ε (IKKε) and the de-ubiquitinase CYLD. This complex forms when fucose signalling through DC-SIGN leads to the disassociation of the RAF1 signalosome and the recruitment of IKKε and CYLD to LSP1. The resultant signalling cascade induces T helper 2 (TH2) and T follicular helper (TFH) cell differentiation.
- Leukotriene
-
A class of eicosanoids derived from the metabolism of arachidonic acid by the action of leukocyte 5-lipoxygenase and other enzymes. They have a conjugated triene double-bond structure and various pro-inflammatory activities, including leukocyte activation (by leukotriene B4) and bronchoconstriction (by leukotriene C4 and leukotriene D4).
Rights and permissions
About this article

Cite this article
Geijtenbeek, T., Gringhuis, S. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol 16, 433–448 (2016). https://doi.org/10.1038/nri.2016.55
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri.2016.55
This article is cited by
-
Nanotheranostic Trojan Horse for visualization and photo-immunotherapy of multidrug-resistant bacterial infection
Journal of Nanobiotechnology (2023)
-
Similar proteome expression profiles of the aggregated lymphoid nodules area and Peyer’s patches in Bactrian camel
BMC Genomics (2023)
-
Vaccine adjuvants: mechanisms and platforms
Signal Transduction and Targeted Therapy (2023)
-
A tumor-associated heparan sulfate-related glycosaminoglycan promotes the generation of functional regulatory T cells
Cellular & Molecular Immunology (2023)
-
Application and prospect of targeting innate immune sensors in the treatment of autoimmune diseases
Cell & Bioscience (2022)
