
Abstract
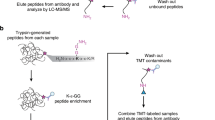
Ubiquitination is essential for the regulation of cellular protein homeostasis. It also has a central role in numerous signaling events. Recent advances in the production and availability of antibodies that recognize the Lys-ɛ-Gly-Gly (K-ɛ-GG) remnant produced by trypsin digestion of proteins having ubiquitinated lysine side chains have markedly improved the ability to enrich and detect endogenous ubiquitination sites by mass spectrometry (MS). The following protocol describes the steps required to complete a large-scale ubiquitin experiment for the detection of tens of thousands of distinct ubiquitination sites from cell lines or tissue samples. Specifically, we present detailed, step-by-step instructions for sample preparation, off-line fractionation by reversed-phase chromatography at pH 10, immobilization of an antibody specific to K-ɛ-GG to beads by chemical cross-linking, enrichment of ubiquitinated peptides using these antibodies and proteomic analysis of enriched samples by LC–tandem MS (MS/MS). Relative quantification can be achieved by performing stable isotope labeling by amino acids in cell culture (SILAC) labeling of cells. After cell or tissue samples have been prepared for lysis, the described protocol can be completed in ∼5 d.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Ye, Y. & Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 (2009).
Dikic, I., Wakatsuki, S. & Walters, K.J. Ubiquitin-binding domains—from structures to functions. Nat. Rev. Mol. Cell Biol. 10, 659–671 (2009).
Peng, J. et al. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 (2003).
Danielsen, J.M.R. et al. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell Proteomics 10, M110.003590 (2010).
Udeshi, N.D. et al. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell Proteomics 11, 148–159 (2012).
Udeshi, N.D. et al. Refined preparation and use of anti-diglycine remnant (K-ɛ-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell Proteomics 12, 825–831 (2013).
Kim, W. et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 (2011).
Xu, G., Paige, J.S. & Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 (2010).
Ong, S.-E. et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376–386 (2002).
Wagner, S.A. et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteomics 10, M111.013284 (2011).
Emanuele, M.J. et al. Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (2011).
Sarraf, S.A. et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature (2013).
Wagner, S.A. et al. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell Proteomics 11, 1578–1585 (2012).
Mertins, P. et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 (2013).
James, G.T. Inactivation of the protease inhibitor phenylmethylsulfonyl fluoride in buffers. Anal. Biochem. 86, 574–579 (1978).
Meng, L. et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo anti-inflammatory activity. Proc. Natl. Acad. Sci. USA 96, 10403–10408 (1999).
Harper, J.W. & Tan, M.-K.M. Understanding cullin-RING E3 biology through proteomics-based substrate identification. Mol. Cell Proteomics 11, 1541–1550 (2012).
Ong, S.E. & Mann, M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 359, 37–52 (2007).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Glatter, T. et al. Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J Proteome Res. 11, 5145–5156 (2012).
Villén, J. & Gygi, S.P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 (2008).
Wang, Y. et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 (2011).
Yang, F., Shen, Y., Camp, D.G. II & Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 9, 129–134 (2012).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Acknowledgements
We thank L. Gaffney for help with illustrations. This work was supported in part by the Broad Institute of MIT and Harvard and by grants from the US National Cancer Institute (U24CA160034, part of the Clinical Proteomics Tumor Analysis Consortium initiative; to S.A.C.) and the National Heart, Lung and Blood Institute (HHSN268201000033C and R01HL096738; to S.A.C.).
Author information
Authors and Affiliations
Contributions
N.D.U., P.M., T.S. and S.A.C. developed the protocol. N.D.U. and S.A.C. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Udeshi, N., Mertins, P., Svinkina, T. et al. Large-scale identification of ubiquitination sites by mass spectrometry. Nat Protoc 8, 1950–1960 (2013). https://doi.org/10.1038/nprot.2013.120
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2013.120
This article is cited by
-
Global ubiquitinome profiling identifies NEDD4 as a regulator of Profilin 1 and actin remodelling in neural crest cells
Nature Communications (2022)
-
Differential ubiquitome analysis of Cordyceps militaris lysine-ubiquitinated proteins affected by blue light
Biologia (2022)
-
Functional significance of gain-of-function H19 lncRNA in skeletal muscle differentiation and anti-obesity effects
Genome Medicine (2021)
-
Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology
Nature Communications (2021)
-
Time-resolved in vivo ubiquitinome profiling by DIA-MS reveals USP7 targets on a proteome-wide scale
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
