
Abstract
The importance of dopamine (DA) neurotransmission is emphasized by its direct implication in several neurological and psychiatric disorders. The DA transporter (DAT), target of psychostimulant drugs, is the key protein that regulates spatial and temporal activity of DA in the synaptic cleft via the rapid reuptake of DA into the presynaptic terminal. There is strong evidence suggesting that DAT-interacting proteins may have a role in its function and regulation. Performing a two-hybrid screening, we identified snapin, a SNARE-associated protein implicated in synaptic transmission, as a new binding partner of the carboxyl terminal of DAT. Our data show that snapin is a direct partner and regulator of DAT. First, we determined the domains required for this interaction in both proteins and characterized the DAT-snapin interface by generating a 3D model. Using different approaches, we demonstrated that (i) snapin is expressed in vivo in dopaminergic neurons along with DAT; (ii) both proteins colocalize in cultured cells and brain and, (iii) DAT and snapin are present in the same protein complex. Moreover, by functional studies we showed that snapin produces a significant decrease in DAT uptake activity. Finally, snapin downregulation in mice produces an increase in DAT levels and transport activity, hence increasing DA concentration and locomotor response to amphetamine. In conclusion, snapin/DAT interaction represents a direct link between exocytotic and reuptake mechanisms and is a potential target for DA transmission modulation.
Similar content being viewed by others


Introduction
Dopaminergic neurons, located primarily in the midbrain, project into various cortical and subcortical regions regulating sensory-motor and cognitive functions. The importance of dopamine (DA) neurotransmission is emphasized by its direct implication in neurological and psychiatric disorders, such as Parkinson’s disease, dystonia, schizophrenia, attention deficit/hyperactivity disorder, Tourette syndrome, and drug addiction (German et al, 2015; Grace, 2016; Martins et al, 2017). These disorders share DA transmission dysfunction as a common pathological mechanism. The key to understand and reverse the pathophysiology of these disorders is to identify molecular and cellular mechanisms that shape the kinetics of DA concentration at synapses (Fon and Edwards, 2001; Lin et al, 2011).
The DA transporter (DAT) is the key protein that removes DA from the synaptic cleft via an ionic gradient-dependent reuptake mechanism (Giros and Caron, 1993). By rapidly clearing DA from the extracellular space, DAT regulates the availability of DA in both time and space. The importance of this reuptake process is sustained by the profound consequences of its blockade using psychostimulant drugs or genetic loss of function (Giros et al, 1996). Indeed, DAT knockout mice display severe behavioral and neurochemical changes, including spontaneous hyperlocomotion, increased DA receptor activation, and paradoxical responses to psychostimulants (Gainetdinov et al, 1999; Giros et al, 1996; Jones et al, 1998).
In light of the accumulating evidence suggesting that DAT-interacting proteins may have a role in its function and regulation (Eriksen et al, 2010), our team performed a yeast two-hybrid (Y2H) screening using the DAT carboxy terminus (DAT-CT) to discover new DAT-associated proteins. In the present study, we report snapin as a new DAT-interacting protein.
Snapin (15 kDa) was first identified as a SNARE-associated protein that binds to SNAP25 and is involved in membrane fusion events (Ilardi et al, 1999). It is ubiquitously expressed and binds SNAP-23, which supports that snapin may control general SNARE-mediated fusion events (Buxton et al, 2003). Snapin participates in synchronizing synaptic vesicle fusion and neurotransmitter release (Pan et al, 2009; Tian et al, 2005). Besides, it coordinates dynein-driven retrograde transport of endosomes and as such, snapin deficiency impairs the delivery of endocytosed material to the endolysosomal system for degradation (Cai et al, 2010; Lee et al, 2012). This role of snapin in endolysosomal transport and sorting has also been applied to the recycling of synaptic vesicles (Di Giovanni and Sheng, 2015). Here we show that snapin is expressed in dopaminergic neurons along with DAT, and that both proteins colocalize in cultured cells and brain. We determined the interaction domains of both proteins, created a 3D model of the complex, and established that snapin modulates DAT uptake activity. Finally, using viral technique to modify gene expression, we demonstrated the relevance of DAT regulation by snapin in vivo.
Materials and methods
Yeast Two-Hybrid
Y2H screening was performed by Hybrigenics (France) and experiments were performed as previously described (De Gois et al, 2015).
Double Labeling Fluorescent In-Situ Hybridization
Hybridization was performed as described (Viereckel et al, 2016). Frozen brain sections of C57BL/6J mice (16 μm) were used. Probes (cRNA) against DAT (nucleotides 1153–2020; GenBank AccNum: NM_012694.2) and snapin (nucleotides 102–1382; GenBank AccNum: NM_133854.3) were generated by in vitro transcription with digogexigenin-dUTP and fluorescein-dUTP, respectively. Slides were scanned with the Nanozoomer Digital Pathology 2.0 high throughput, its fluorescence unit option (L11600-05), and the NanoZoomer’s 3-CCD time delay integration camera (Hamamatsu Photonics). Resolution of 0.23 μm/pixel (× 40) (or 0.46 μm/pixel (× 20)) was used.
Immunocytochemistry
Forty-eight hours after transfection, cells were rinsed with PBS then fixed on ice with 4% paraformaldehyde for 10 min. Cells were rinsed three times for 10 min with PBS and incubated 30 min at room temperature with blocking buffer (0.2% Triton X-100 and 3% normal goat serum in PBS). Animals were perfused with 4% paraformaldehyde in PBS and brains were cut in 40 μm-thick slices on a vibrating microtome (Leica Biosystems, VT1000S). Primary antibodies anti-DAT (Chemicon, 1:1000) and anti-Snapin (Synaptic Systems, 1:1000) were incubated overnight at 4 °C. After three washes for 10 min with PBS, secondary antibodies coupled to Alexa 488 or 555 (Life Technologies, 1:2000) were applied for 1 h at room temperature. Nuclei were labeled using DAPI (Sigma-Aldrich, 1:20 000). Glass coverslips were mounted on a slide with Fluoromount-G (Clinisciences). All images were acquired with a × 63 oil-immersion objective on confocal laser-scanning microscope (Leica TCS SP5 with LCS Leica software) using a z-stack of 0.5 μm intervals.
Proximity Ligation Assay
Experiments were performed according to the manufacturer’s instructions with the following modifications: proximity ligation assay (PLA) probes incubation was for 2 h, ligation was performed for 45 min, and amplification step was extended for 2 h with a concentration of polymerase of 1/60 (all steps at 37 °C). BON cultured cells were plated into eight-well LabTek slides (Dutscher). Blocking (1 h at room temperature) and primary antibody (overnight at 4 °C) incubations were performed in a 3% bovine serum albumin and 0.2% Triton X-100 solution. Rabbit anti-Snapin (Synaptic Systems) and rat anti-DAT (Chemicon) were diluted (1:1000) in the blocking solution, as well as the anti-rabbit (+) PLA probe (1:5) along with an anti-rat (−) probe (1:100). Anti-rat PLA probes were generated according to the manufacturer’s instructions using the Duolink Probemaker (Olink Bioscience). Goat anti-rat IgGs (Santa Cruz) were used as control. Slides were mounted in Fluoromont-G. Images were acquired as above.
Biotinylation
Biotinylation was performed using the Cell Surface Protein Isolation Kit (Pierce) according to the manufacturer’s protocol, with 0.5 mg of sulfo-NHS-SS-biotin for 0.5 mg of synaptosomes.
Western Blot Analysis
Cells were rinsed with PBS, collected on ice by scraping in cold PBS containing antiproteases, sonicated, and centrifuged at 2000 g for 10 min at 4 °C. Protein concentration of the post-nuclear supernatant was determined with a Bradford assay (Bio-Rad). Proteins were size fractionated on a 10% precast Bis-Tris polyacrylamide gel (Life Technologies) and electrophoretically transferred to a nitrocellulose membrane (GE Healthcare) using standard protocols. Membranes were blocked for 1 h at room temperature (in 5% non-fat dry milk in PBS/Tween 0.1%), then incubated overnight at 4 °C with primary antibodies directed against DAT (Chemicon, 1:1000), snapin (Synaptic Systems 1:1000), Myc (Clontech, 1:250), glyceraldehyde 3-phosphate dehydrogenase (Calbiochem, 1:5000), or actin (Sigma, 1:5000). Fluorescent secondary antibodies were detected and quantified using the Odyssey infrared imaging system (LI-COR Biosciences). For DAT, horseradish peroxidase-conjugated secondary antibodies were detected with enhanced chemiluminescence using an ImageQuant LAS 4000 Imager.
Striatal Synaptosomes
Mouse or rat brains were collected in ice-cooled dishes. Striata were homogenized in ice-cold 0.32 M sucrose using a Teflon-glass homogenizer, centrifuged at 1000 g for 10 min at 4 °C. The resulting supernatant was centrifuged at 12 000 g for 15 min. Pellet was washed in 0.32 M sucrose and finally suspended in the appropriate buffer.
DA Uptake on Synaptosomes
Uptake were performed as described (De Gois et al, 2015) with some modifications. A MultiScreen HTS vacuum manifold with 96-well plates (Merck Millipore) were used with 10–15 μg of striatal synaptosomes. Thirty nanomolar of 3H-DA (Perkin Elmer) was used with increasing concentration of unlabeled substrate (70–2970 nM) for the characterization of DAT kinetic parameters.
Viral Manipulation and Animal Surgery
Snapin shRNA (mouse) lentiviral particles (sc-45546-V, Santa Cruz Biotechnology, USA) contain 3-5 expression constructs each encoding target-specific 19–25 nucleotides (plus hairpin) shRNA designed to knockdown snapin gene expression. Control shRNA lentiviral particles (sc-108080) encode a scrambled shRNA sequence. For stereotaxic delivery of the viruses, mice were anesthetized with a ketamine/xylazine mixture (100/10 mg/kg, i.p.) and then given bilateral microinjections (1.0 μl per side over 10 min) into the substantia nigra pars compacta (SNc) (anteroposterior, −3.1 mm; lateral, +1.7 mm; dorsoventral, −4.4 mm below dura), using a 32-gauge Hamilton syringe angled at 7°.
Locomotor Activity
Mice were introduced into a circular corridor (4.5 cm width, 17 cm external diameter) crossed by four infrared beams (1.5 cm above the base) placed at every 90° (Imetronic, Pessac). Locomotor activity was scored when animals interrupted two successive beams and thus had traveled one-quarter of the circular corridor. Spontaneous activity was recorded for 60 min. Mice were first injected with saline and 60 min later with amphetamine (3 mg/kg). Locomotor responses were recorded for an additional 120 min.
Additional information about these methods and other methods are available in the Supplementary Information.
Results
Interaction of DAT and Snapin in the Y2H System and in silico
To search for DAT-interacting proteins, a Y2H screening was performed using a rat brain library and the intracellular DAT-CT as bait. From several million transformants screened, 16 positive clones were found to encode sequences of the open reading frame of snapin (411 bp). All clones contained the central large contig 43–364 bp. To rule out the possibility of a false positive interaction, we tested several control constructs in Y2H. Only transformants bearing the bait plasmid pB6-DAT-CT and the prey plasmid pP6-Snapin were positive for the β-galactosidase and the histidine selection (Supplementary Figure S1a). We also examined the specificity of this interaction by determining the ability of snapin to bind to the norepinephrine transporter (NET), a closely related SLC6a family member. We found that snapin also interacted with the intracellular NET-CT (Supplementary Figure S1a).
To determine which regions of DAT are involved in the formation of the DAT/snapin complex, we assessed the ability of various portions of DAT-CT to interact with snapin in Y2H. As illustrated in Figure 1a, DAT/snapin complex formation depends on twelve residues of DAT-CT (Cys580 to Ala591). Indeed, deletion of the last 28 or the first 6 amino acids of DAT-CT does not interfere in snapin interaction with DAT, whereas the deletion of the last 30 residues of DAT-CT results in the loss of this interaction. To more precisely define the critical amino acids required, we inserted mutations in these twelve residues of DAT-CT (Figure 1b). However, single or several multiple mutations in this sequence (or the entire DAT-CT, Supplementary Figure S1b) had no effect on DAT/snapin interaction, thus suggesting that a particular combination of several amino acids within DAT-CT is necessary for this interaction.

Mapping the interaction interface for snapin/dopamine transporter (DAT) complex. (a, b) Schematic representations of the carboxyl tail of DAT (DAT-CT) in yeast 2-hybrid assays (Y2H). Bait cDNAs were fused to the GAL4 binding (G4BD). (a) Y2H assay with truncated DAT-CT reveals the importance of a sequence in the first half of DAT-CT (Cys581–Ala592) for the interaction with snapin. (b) Single or multiple mutations in the FREKLAYA motif of DAT-CT does not impair DAT interaction with snapin. (c, d) Schematic representations of snapin in Y2H assays. Prey cDNAs were fused to the GAL4 activation domains (G4AD). (c) Y2H assay with truncated snapin, based on the predicted 3D structure, reveals that the presence of both helical domains (HD1 and HD2) are required for the interaction with DAT. (d) Y2H assay based on our new predicted structure of snapin reveals that HD1A, HD1B, and HD2A, but not HD2B, are required for the formation of DAT/snapin complex. (e, f) Three-dimensional modeling of the snapin/DAT complex. (e) Upper panel shows DAT primary sequence. Purple boxes overlap amino acids of DAT transmembrane domains 2 and 3 (first lane), 10 and 11 (middle lane), and 12 (lower lane). Black boxes highlight the residues that interact with snapin. Those are mainly located in the CT, but also in the internal loops 2–3 and 10–11. Lower panel represents a partial sequence of snapin where black boxes pointed to the residues required for the interaction with DAT. (f) Representation of snapin (gold) interacting with DAT-CT (green) using DS Modeling 4.5 software, with a higher magnification in the right panel.
Next, we studied which regions of snapin are involved in the DAT/snapin complex. Two helical domains (HDs) have been predicted in snapin (Buxton et al, 2003): HD1 residues 37–65 and HD2 residues 81–126. We observed that deletion mutants of snapin lacking either HD1 or HD2 do not bind to DAT-CT (Figure 2a and Supplementary Figure S2a). This suggested that both HD are necessary for the interaction, and the smallest positive contig was 37–126. Based on the recently crystallized hemoglobin of Cormorant (PDB 3WR1), we generated a new prediction for snapin 3D structure. According to our model, each putative HD of snapin would be subdivided in two, resulting in a total of four HD. We thus generated additional mutants based on this new structure and found that the second part of the HD2 (HD2B) is not required for DAT/snapin interaction (Figure 1d and Supplementary Figure S1d). This helped us further restrict the smallest positive contig to residues 35–99. We also evaluated point mutations of Ser-50 and Cys-66 in snapin, which have been reported to modulate protein phosphorylation, structure, and stability (Chheda et al, 2001; Navarro et al, 2012). We found out that snapin S50A, mimicking the unphosphorylated protein, no longer interacted with DAT, whereas S50D, emulating the phosphorylated state, still did (data not shown). To further characterize the binding interface between DAT-CT and snapin, we generated a three-dimensional model (Figure 1e). The primary step in the generation of a homology model is the sequence alignment between the protein of interest and a crystallized protein. We previously created an homology model for DAT (De Gois et al, 2015) that we refined following publications of two new DAT crystallized structure (Penmatsa et al, 2015; Wang et al, 2015). For snapin, the model was based on the hemoglobin of the Cormorant. After model generation (50 models per protein) and selection (energy-based), we processed the docking of the best homology model of snapin to that of DAT-CT, which was used as a receptor. We generated ~2000 poses clustered according to their distances and interaction energies. The most representative clusters were analyzed and highlighted the interaction on a specific portion of DAT-CT. We selected the best pose in these clusters according to the quality of the complex structure (Figure 1e and Supplementary Figure S1e). The results confirmed that snapin interacts with DAT-CT. More precisely, around half of the residues in snapin (from 36 to 118, mainly located in HD1A, HD1B and HD2A) participate in the interaction (Figure 1d). As for DAT, the 3D model showed that almost the entire DAT-CT (from L582 to the end) is involved in the interaction with snapin (Figure 1d). This suggests that even if only some residues in the Cys580–Ala591 sequence are essential for the direct interaction, many other amino acids may participate in stabilizing the complex.

Coexpression and colocalization of dopamine transporter (DAT) and snapin in brain and cells. (a) Double-labeling fluorescent in situ hybridization (FISH) in mouse brain. Lower panel is a magnification of the white square of the upper panel. FISH showed that snapin mRNAs (labeled with fluorescein, in green) are expressed in all DAT-positive neurons (labeled with digoxigenin, in red) (arrowheads). Some snapin-positive and DAT-negative neurons are also visible (arrows). Scale bar: 250 μm. (b) Immunocytochemistry (ICC) shows that snapin colocalizes with DAT in vitro in cotransfected BON cells, particularly in cell processes. Scale bar: 10 μm. (c) Immunohistochemistry (IHC) confirms DAT and snapin colocalization in mouse brain neurons. Scale bar: 20 μm. The white arrows denote the colocalization of snapin and DAT. (d, e) Duolink proximity ligation assays (PLAs) show that snapin and DAT interact in (d) cotransfected cells, and in (e) mouse brain striatum (note that the signal is absent in the corpus callosum (Cc) where there is no DAT, and in the DAT knockout mice striatum). The arrow in d points to a contransfected cell in which all the red dots are indicative of positive PLA signal. Scale bars: 10 μm.
Coexpression and Colocalization of DAT and Snapin In Vitro and In Vivo
We investigated whether DAT and snapin transcripts are coexpressed in neurons using double-labeling fluorescent in-situ hybridization (FISH). As previously described (De Gois et al, 2015; Giros et al, 1991), DAT expression is restricted to the SNc and the ventral tegmental area (VTA), whereas snapin expression profile is more ubiquitous (Figure 2a and Supplementary Figure S2a). At high magnification, we observed that snapin and DAT transcripts are coexpressed in neurons of the SNc and VTA (Figure 2a). We next examined whether these two proteins colocalized in mammalian cells by coexpressing snapin (mouse) and DAT in BON cells that possess the machinery for a proper expression of active transporters (Tran et al, 2004). As expected, the DAT protein is detected at the plasma membrane with some labeling in the cytoplasm, whereas snapin displayed a widespread cytoplamic expression. Interestingly, snapin colocalizes with DAT, particularly in cell processes (Figure 2b). Furthermore, we established that endogenous DAT and snapin proteins colocalize in dopaminergic nigrostriatal fibres in vivo in the mouse (Figure 2c).
To demonstrate that DAT and snapin belong to the same protein complex we used PLAs to show the physical proximity of DAT and snapin in cells and mouse brain neurons. The PLA assays showed a strong punctate signal in cells coexpressing snapin and DAT, and in DA neurons in the striatum of wild-type mice. Conversely, no PLA signal was detected in DAT knockout animals or in regions where DAT is not expressed (Figures 2d and e, and Supplementary Figure S2). Moreover, we performed a Myc-tagged DAT immunoprecipitation resulting in the co-precipitation of snapin, only when both proteins are coexpressed in BON cells (Figure 3a). In addition, GST-pulldown assays showed that GST-snapin precipitated DAT from striatal synaptosomes of wild-type mice (Figure 3b). As controls, we showed that GST-snapin also precipitated its well-established partner Snap25, but that DAT was not co-precipitated when using DAT knockout mice or GST alone. At last, immunoprecipitation assays in mouse brain confirmed this complex formation in vivo, as DAT and snapin selectively co-immunoprecipitated from striatal synaptosomes (Supplementary Figure S3c). Overall, these data establish the physical interaction between DAT and snapin.

Snapin and dopamine transporter (DAT) are present in the same protein complex and snapin regulates DAT activity. (a) Immunoprecipitation assays demonstrate that DAT forms a complex with snapin in cells coexpressing both proteins. Immunoblot analysis indicated that Myc-tagged DAT immunoprecipitation (with a DAT antibody) resulted in the co-precipitation of snapin when both proteins are present in the cell. Myc-tagged DAT was detected in the immunoblot with a Myc antibody. (b) GST-pull down experiments demonstrate that GST-snapin precipitated DAT from striatal synaptosomes of wild-type mice. In control studies, DAT was not detected in DAT knockout mice or using GST alone; GST-snapin also precipitated Snap25. (c) DAT-mediated [3H]-DA uptake was significantly decreased in cells cotransfected with the DAT and snapin. The graph displays the means ± SEM of six separate experiments performed in triplicate. (d) This effect disappeared when siRNA against snapin was included in the costransfection. The graph displays the means ±SEM of three separate experiments performed in triplicate.
Effect of Snapin on DAT Uptake Activity In Vitro
To identify whether this direct interaction may have any functional relevance, we tested the effect of snapin expression on DAT activity in cotransfected cells. Snapin overexpression dramatically decreased DAT-mediated DA uptake by 40% of the maximal velocity (Vmax), with no changes in the KM value (Vmax: 100.0±2.5% for DAT, 63.1±1.9% for DAT+Sn; Km (nM): 2167±194 for DAT, 1866±214 for DAT+Sn; different curve for each data set, F(2,349)=156.0, p<0.0001; with differences in the Vmax, F(1,349)=128.1, p<0.0001) (Figure 3c). The downregulation of DA uptake was specifically mediated by snapin overexpression, as this effect was abolished when cells were cotransfected with a siRNA targeting the exogenous snapin (F(2,172)= 0.6625, p=0.5168, NS; Figure 3d).
Functional Consequences of the DAT/Snapin Interaction In Vivo
To test the functional consequences of decreased snapin expression in vivo, we injected a lentivirus expressing shRNA targeting snapin or control in the SNc and evaluated the consequences of such manipulations over DAT expression in the striatal DA terminals ascending from the SNc. As a preliminary step, we checked in vitro that the siRNA sequences, which were used to produce the lentivirus, completely silenced the expression of snapin in transfected cells, without altering the expression of other proteins (Supplementary Figure S3). In vivo, snapin shRNA injection in the SNc resulted in a marked 23% knockdown of snapin levels in the striatum (Supplementary Figure S3). This may correspond to a strong donwregulation of the snapin located on the striatal terminals projecting from the SNc (at least 50%), whereas snapin in the striatal neuronal cell bodies are not affected.
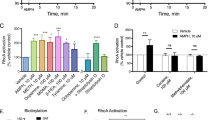
Snapin shRNA injected-mice showed a dramatic increase (40–70%) in DAT expression in the striatal terminals, both at the membrane and intracellular levels. As shown in Figure 4a, western blot and biotinylation studies revealed an increase in DAT levels in the total (*p=0.0211), biotinylated membranous (*p=0.0211) and non-biotinylated intracellular fractions (*p=0.0441). Electron microscopy analysis confirmed this augmentation in DAT levels in the striatal terminals, although the difference reached significance only in the intracellular DAT located in the terminals (*p=0.0454; Figure 4b). Accordingly, snapin downregulation produced a significant increase in DAT-mediated DA uptake (~ +40% of the Vmax, with no changes in the affinity) in striatal synaptosomes of these mice (different curve for each data set, F(2,124)=8.491, p=0.0003; with differences in the Vmax, F(1,124)=9.297, p=0,0028; Figure 4c).

Snapin downregulation in mouse brain results in an increase in DAT protein levels and activity. (a) Western blotting of striatal synaptosomes, along with the biotinylated and non-biotinylated fractions, show an increase in DAT levels in snapin shRNA-injected animals in the total, biotinylated membranous and non-biotinylated intracellular fractions. The bars show the means ±SEM of eight animals performed in duplicate. (b) Electron microscopy analysis and quantification confirmed this increase in DAT levels in the stiatal terminals. The bars show the means ±SEM of six animals. Eight to twenty terminals were quantified per animal. (c) Increased DAT mediated [3H]-DA uptake in striatal synaptosomes in snapin shRNA-injected animals. The graph displays the means ±SEM of four separate experiments performed in triplicate.
As DAT activity in striatum has a major role in locomotion and psychostimulant sensitivity, we studied the behavioral consequences of manipulating snapin on amphetamine-induced locomotor activity. Snapin shRNA injected mice presented a considerably increased locomotor response to amphetamine (3 mg/kg), with no differences in basal activity (FD[1,348]=10.96, p=0.0162; Ft[58,348]=33.78, p<0.0001; Fi[58,348]=2.658, p<0.0001; Figure 5a). No changes were observed in the stereotypies produced by amphetamine at this dose, which could have hampered locomotion (p=0.6616, NS; Figure 5b). Given that amphetamine does not only inhibit DAT, but can also directly trigger DA release through reversal of the DAT uptake activity, these behavioral results agree with the higher DAT levels observed. This increase in amphetamine-triggered locomotor activity is highly correlated to the strong augmentation (+60 %) in extracellular levels of DA after amphetamine in snapin shRNA animals, as directly measured by microdialysis performed on freely moving animals (FD[1,88]=7.617, p=0.0247; Ft[11,88]=71.43, p<0.0001; Fi[11,88]=5.468, p<0.0001; Figure 5c), which showed no difference in the basal DA levels (p=0.9410, NS; Figure 5d). Moreover, amphetamine effect was absent when snapin shRNA was injected in SNc of DAT knockout mice (Supplementary Figure S4).

Snapin downregulation in mouse brain produces strong behavioral and neurochemical consequences. (a) Increased locomotor activity after amphetamine (3 mg/kg) injection was observed after snapin shRNA mice. The graph displays the means ±SEM of four animals. (b) Increased extracellular dopamine (DA) concentration measured with microdialysis in control and snapin shRNA injected mice after amphetamine (3 mg/kg) injection. (c,d) Snapin shRNA-injected mice showed no significant differences in the locomotor response to cocaine or the extracellular striatal DA concentration in response to cocaine injection (10 mg/kg), as measured by microdialysis performed on freely moving animals. The graph displays the means ±SEM of four animals. (e) Snapin shRNA-injected mice showed no significant differences in the microdialysis measurement of striatal DA release evoked by potassium (50 mM) in control and snapin shRNA injected mice. The graph displays the means ±SEM of seven animals.
In contrast, when the same experiments were performed with cocaine (10 mg/kg), a ‘pure’ DAT blocker with no releasing activity, we did not observe any significant difference between animals, either in the cocaine-induced locomotor activity (FD[1,312]=4.182, p=0.0617; Ft[24,312]=69.72, p<0.0001; Fi[24,312]=0.5888, p=0.9395) or cocaine-induced extracellular DA levels (FD[1,66]=0.1010, p=0.7614; Ft[11,66]=13.89, p<0.0001; Fi[11,66]=0.4462, p=0.9287; Figures 5e and f). Finally, to make sure that this was not a ‘per se’ effect of snapin downregulation following shRNA administration, we assessed that the K+-evoked vesicular DA release was unchanged in snapin shRNA-injected mice (FD[1,120]=0.063, p=0.8060; Ft[10,120]=13.21, p<0.0001; Fi[10,120=0.1488, p=0.9988; Figure 5g).
Discussion
Herein, we characterized snapin as a novel DAT partner and regulator. We showed that DAT and snapin colocalize and directly interact in cells and neurons. We also established that snapin downregulates DAT expression and uptake activity and demonstrated the relevance of this regulation in mice.
Role of Snapin in DAT Regulation
One of the recent roles attributed to snapin is the coordination of endolysosomal retrograde transport and sorting, which has also been applied to the recycling of synaptic vesicles (Cai et al, 2010; Di Giovanni and Sheng, 2015). As snapin deficiency impairs the delivery of endocytosed material to the endolysosomal system for degradation (Cai et al, 2010; Di Giovanni and Sheng, 2015), snapin downregulation could increase DAT levels by reducing its degradation. Given that we report a direct interaction between snapin and DAT, this could suggest that the formation of snapin/DAT complex is necessary for targeting the recycling DAT-containing vesicles towards the endocytic pathways. Another possible mechanism for the regulation of DAT by snapin could involve TorsinA, which regulates DAT trafficking (Torres et al, 2004) and also interacts with snapin, having a role in regulated exocytosis (Granata et al, 2008, 2011). At last, vesicle endo/exocytose implicates various SNARE and related proteins (Südhof, 2013), some of which interact with snapin, such as Snap25 (Ilardi et al, 1999) and synaptotagmin (Chheda et al, 2001), or with DAT such as Syntaxin1A (Binda et al, 2008; Carvelli et al, 2008; Lee et al, 2004) and Synpatogyrin-3 (Egaña et al, 2009). The formation of snapin/DAT could thus contribute to an increased endocytosis or decreased export of DAT-containing vesicles. However, it remains unknown whether the formation of DAT/snapin complexes is constitutive or regulated by neural activity, and whether it occurs at a specific stage in the transporter life cycle.
We show that DAT/snapin complex formation depends on twelve amino acids in the carboxyl terminal of DAT (Cys580–Ala591). It has already been suggested that the central motif FREKLAYAIA (residues 587–596) in hDAT is essential for the constitutive internalization of the transporter (Holton et al, 2005). Importantly, this motif overlaps with the domain we found necessary for the interaction with snapin. This suggests that snapin might regulate the internalization of DAT through this domain. However, Holton et al (2005) showed that point mutations in several residues in the FREKLAYAIA motif (L591A, Y593A and I595A in hDAT, being L590, Y592 and I594 in rat) still expressed a similar distribution to that of wild-type DAT. Accordingly, we observed that point mutations are not sufficient to interrupt the interaction suggesting that a particular combination of several amino acids within this region is necessary for the interaction. In addition, Miranda et al (2004) found that alanine substitution of K590 in hDAT (K589 in rat) resulted in a strong intracellular localization, produced by a significant delay, not a complete blockade, of the delivery of DAT to the plasma membrane because of its retention in the endoplasmic reticulum. This is in agreement with our results of single or multiple mutations of K589, which still interact with snapin. At last, another study showed that progressive deletions in DAT-CT produced a progressive decrease in transport activity and export to the membrane (Torres et al, 2003). More precisely, a DAT-CT mutant stopping at L591 (corresponding to our DAT-ΔCT30) exhibited less than 1% of wild-type uptake function, and a mutant stopping at S582 completely abolished transporter function (Torres et al, 2003). This suggests that mutants that cannot bind snapin are not correctly exported to the membrane, again reinforcing the hypothesis that snapin has a role in DAT trafficking. Other studies identifying the molecular determinants regulating transporter trafficking and interactions with other proteins have focused on domains in DAT-CT others than the one identified in our study: the PDZ domain (Rickhag et al, 2013; Torres et al, 2001), residues 612–617 flanking the PDZ domain (Bjerggaard et al, 2004; Fog et al, 2006), and the initial residues before C580 (Carneiro et al, 2002; De Gois et al, 2015).
New Insights into the Structure of Snapin
Another interesting aspect of our work is that we provided new insights into the structure of snapin. Two HD were initially predicted in snapin (Buxton et al, 2003). A posterior bioinformatic study that modeled the potential coiled-coil structure of snapin also proposed two helicodial domains composed of similar series of residues (Gowthaman et al, 2006). We could notice that secondary structure prediction of these types of structure remained challenging. Indeed, various prediction tools such as DSC (King and Sternberg, 1996), Predictprotein (Yachdav et al, 2014), or YASPIN (Lin et al, 2005) failed to accurately predict the secondary structure of putative crystallized templates (pdb: 3WR1_A;15MI; 3DHR). Based on this observation, we selected the best template based on sequence similarity and identity. Thus, we generated a new prediction of the 3D structure for snapin, based on the recently crystallized hemoglobin (PDB code: 3WR1), which has higher sequence homology and identity with snapin (Identity 25%; Similarity: 50% over 92AA). According to our model, each of the putative HD of snapin would be separated in two, resulting in a total of four HD. With mutants based on this new structure and we observed that the second part of the HD2 is not required for the interaction with DAT, which could be considered as a validation of the new structure.
The Functional Interaction Between Snapin and DAT Differentially Modulates Psychostimulant Actions
We provide strong evidence for the relevance of DAT regulation by snapin in vivo: snapin downregulation in mice increases DAT levels and activity, hence increasing DA concentration and locomotor response to amphetamine. It is striking to see that the phenotype induced by decreasing the snapin/DAT interaction fully recapitulates the one observed in animals overexpressing DAT (Calipari et al, 2013, 2015; Salahpour et al, 2008). Indeed, animals overexpressing DAT display a threefold increase in the amount of DA released by amphetamine, compared with controls, that correlates with a threefold increase in protein expression of total and membrane DAT. Behaviorally, these mice also present a marked increase in locomotor responses to amphetamine compared with WT animals (Salahpour et al, 2008). Interestingly, DAT blockers such as cocaine, GBR12909 and methylphenidate induced similar locomotor activities in these mice and WT animals (Salahpour et al, 2008). It has also been shown that transgenic animals overexpressing DAT display enhanced neurochemical potency and reinforcing effects of amphetamine, but not DAT blockers (Calipari et al, 2013, 2015). The same findings have been reported in a rat model of methylphenidate self-administration, which causes elevations in DAT levels; these rats show enhanced potency for amphetamine on DA responses and drug seeking behaviors, without altered cocaine effects (Calipari et al, 2013, 2015). We observed the same dissimilarity in mice with snapin downregulation. These mice showed increased DA concentration and locomotor response after amphetamine administration but displayed no significant differences in cocaine-triggered responses. It thus seems that the effects of amphetamine at the dopaminergic terminal are dependent and directly proportional to the levels of DAT, while the effects of blockers such as cocaine are substantially less susceptible to DAT overexpression.
To conclude, our data demonstrate that the newly identified snapin/DAT interaction has an important relevance in vivo: snapin downregulation directly increases DAT expression and activity at the membrane. This functional interaction represents a novel and direct link between exocytotic and reuptake organizational clusters that may have a direct impact on our understanding of synaptic functions in the normal brain and synaptic deficits in various psychiatric disorders. Thus, this novel snapin/DAT complex represents a potential target for DA transmission modulation, essential to many neuropsychiatric diseases.
Funding and disclosure
AME was recipient of a postdoctoral fellowship from Fondation pour la Recherche Médicale (2011–2012) and from the Basque Government (2013–2015). This work was funded by Canadian Institutes of Health Research (to BG), Schizo Oui Association, and the Instituto de Salud Carlos III, Centro de Investigación Biomédica en Red de Salud Mental, CIBERSAM, Spain. The authors declare no conflict of interest.
References
Binda F, Dipace C, Bowton E, Robertson SD, Lute BJ, Fog JU et al (2008). Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Mol Pharmacol 74: 1101–1108.
Bjerggaard C, Fog JU, Hastrup H, Madsen K, Loland CJ, Javitch JA et al (2004). Surface targeting of the dopamine transporter involves discrete epitopes in the distal C terminus but does not require canonical PDZ domain interactions. J Neurosci 24: 7024–7036.
Buxton P, Zhang XM, Walsh B, Sriratana A, Schenberg I, Manickam E et al (2003). Identification and characterization of Snapin as a ubiquitously expressed SNARE-binding protein that interacts with SNAP23 in non-neuronal cells. Biochem J 375: 433–440.
Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH (2010). Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68: 73–86.
Calipari ES, Ferris MJ, Salahpour A, Caron MG, Jones SR (2013). Methylphenidate amplifies the potency and reinforcing effects of amphetamines by increasing dopamine transporter expression. Nat Commun 4: 2720.
Calipari ES, Ferris MJ, Siciliano CA, Jones SR (2015). Differential influence of dopamine transport rate on the potencies of cocaine, amphetamine, and methylphenidate. ACS Chem Neurosci 6: 155–162.
Carneiro AM, Ingram SL, Beaulieu J-M, Sweeney A, Amara SG, Thomas SM et al (2002). The multiple LIM domain-containing adaptor protein Hic-5 synaptically colocalizes and interacts with the dopamine transporter. J Neurosci 22: 7045–7054.
Carvelli L, Blakely RD, DeFelice LJ (2008). Dopamine transporter/syntaxin 1A interactions regulate transporter channel activity and dopaminergic synaptic transmission. Proc Natl Acad Sci USA 105: 14192–14197.
Chheda MG, Ashery U, Thakur P, Rettig J, Sheng ZH (2001). Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat Cell Biol 3: 331–338.
De Gois S, Slama P, Pietrancosta N, Erdozain AM, Louis F, Bouvrais-Veret C et al (2015). Ctr9, a protein in the transcription complex Paf1, regulates dopamine transporter activity at the plasma membrane. J Biol Chem 290: 17848–17862.
Di Giovanni J, Sheng Z-H (2015). Regulation of synaptic activity by snapin-mediated endolysosomal transport and sorting. EMBO J 34: 2059–2077.
Egaña LA, Cuevas RA, Baust TB, Parra LA, Leak RK, Hochendoner S et al (2009). Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-3. J Neurosci 29: 4592–4604.
Eriksen J, Jorgensen TN, Gether U (2010). Regulation of dopamine transporter function by protein-protein interactions: new discoveries and methodological challenges. J Neurochem 113: 27–41.
Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N et al (2006). Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51: 417–429.
Fon EA, Edwards RH (2001). Molecular mechanisms of neurotransmitter release. Muscle Nerve 24: 581–601.
Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG (1999). Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science 283: 397–401.
German CL, Baladi MG, McFadden LM, Hanson GR, Fleckenstein AE (2015). Regulation of the dopamine and vesicular monoamine transporters: pharmacological targets and implications for disease. Pharmacol Rev 67: 1005–1024.
Giros B, Caron MG (1993). Molecular characterization of the dopamine transporter. Trends Pharmacol Sci 14: 43–49.
Giros B, El Mestikawy S, Bertrand L, Caron MG (1991). Cloning and functional characterization of a cocaine-sensitive dopamine transporter. FEBS Lett 295: 149–154.
Giros B, Jaber M, Jones SR, Wightman RM, Caron MG (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379: 606–612.
Gowthaman R, Silvester AJ, Saranya K, Kanya KS, Archana NR (2006). Modeling of the potential coiled-coil structure of snapin protein and its interaction with SNARE complex. Bioinformation 1: 269–275.
Grace AA (2016). Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci 17: 524–532.
Granata A, Koo SJ, Haucke V, Schiavo G, Warner TT (2011). CSN complex controls the stability of selected synaptic proteins via a torsinA-dependent process. EMBO J 30: 181–193.
Granata A, Watson R, Collinson LM, Schiavo G, Warner TT (2008). The dystonia-associated protein torsinA modulates synaptic vesicle recycling. J Biol Chem 283: 7568–7579.
Holton KL, Loder MK, Melikian HE (2005). Nonclassical, distinct endocytic signals dictate constitutive and PKC-regulated neurotransmitter transporter internalization. Nat Neurosci 8: 881–888.
Ilardi JM, Mochida S, Sheng ZH (1999). Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat Neurosci 2: 119–124.
Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG (1998). Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA 95: 4029–4034.
King RD, Sternberg MJ (1996). Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci 5: 2298–2310.
Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC et al (2012). Assembly and architecture of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J Biol Chem 287: 5882–5890.
Lee K-H, Kim M-Y, Kim D-H, Lee Y-S (2004). Syntaxin 1A and receptor for activated C kinase interact with the N-terminal region of human dopamine transporter. Neurochem Res 29: 1405–1409.
Lin K, Simossis VA, Taylor WR, Heringa J (2005). A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics 21: 152–159.
Lin Z, Canales JJ, Björgvinsson T, Thomsen M, Qu H, Liu Q-R et al (2011). Monoamine transporters: vulnerable and vital doorkeepers. Prog Mol Biol Transl Sci 98: 1–46.
Martins D, Mehta MA, Prata D (2017). The “highs and lows” of the human brain on dopaminergics: Evidence from neuropharmacology. Neurosci Biobehav Rev 80: 351–371.
Miranda M, Sorkina T, Grammatopoulos TN, Zawada WM, Sorkin A (2004). Multiple molecular determinants in the carboxyl terminus regulate dopamine transporter export from endoplasmic reticulum. J Biol Chem 279: 30760–30770.
Navarro A, Encinar JA, López-Méndez B, Aguado-Llera D, Prieto J, Gómez J et al (2012). Mutation of Ser-50 and Cys-66 in Snapin modulates protein structure and stability. Biochemistry 51: 3470–3484.
Pan PY, Tian JH, Sheng ZH (2009). Snapin facilitates the synchronization of synaptic vesicle fusion. Neuron 61: 412–424.
Penmatsa A, Wang KH, Gouaux E (2015). X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat Struct Mol Biol 22: 506–508.
Rickhag M, Hansen FH, Sørensen G, Strandfelt KN, Andresen B, Gotfryd K et al (2013). A C-terminal PDZ domain-binding sequence is required for striatal distribution of the dopamine transporter. Nat Commun 4: 1580.
Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E et al (2008). Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc Natl Acad Sci USA 105: 4405–4410.
Südhof TC (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80: 675–690.
Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C et al (2005). The role of Snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci 25: 10546–10555.
Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A, Yao W-D et al (2003). Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J Biol Chem 278: 2731–2739.
Torres GE, Sweeney AL, Beaulieu J-M, Shashidharan P, Caron MG (2004). Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci USA 101: 15650–15655.
Torres GE, Yao WD, Mohn AR, Quan H, Kim KM, Levey AI et al (2001). Functional interaction between monoamine plasma membrane transporters and the synaptic PDZ domain-containing protein PICK1. Neuron 30: 121–134.
Tran VS, Marion-Audibert A-M, Karatekin E, Huet S, Cribier S, Guillaumie K et al (2004). Serotonin secretion by human carcinoid BON cells. Ann N Y Acad Sci 1014: 179–188.
Viereckel T, Dumas S, Smith-Anttila CJA, Vlcek B, Bimpisidis Z, Lagerström MC et al (2016). Midbrain gene screening identifies a new mesoaccumbal glutamatergic pathway and a marker for dopamine cells neuroprotected in Parkinson’s disease. Sci Rep 6: 35203.
Wang KH, Penmatsa A, Gouaux E (2015). Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 521: 322–327.
Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T et al (2014). PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 42: W337–W343.
Acknowledgements
We thank David Godefroy (Plateform of Institut de la Vision, Paris, France) for his technical help in acquiring the scans of the FISH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Erdozain, A., De Gois, S., Bernard, V. et al. Structural and Functional Characterization of the Interaction of Snapin with the Dopamine Transporter: Differential Modulation of Psychostimulant Actions. Neuropsychopharmacol. 43, 1041–1051 (2018). https://doi.org/10.1038/npp.2017.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.217
This article is cited by
-
Identification by proximity labeling of novel lipidic and proteinaceous potential partners of the dopamine transporter
Cellular and Molecular Life Sciences (2021)
-
Cartography of hevin-expressing cells in the adult brain reveals prominent expression in astrocytes and parvalbumin neurons
Brain Structure and Function (2019)
-
Melatonin receptors limit dopamine reuptake by regulating dopamine transporter cell-surface exposure
Cellular and Molecular Life Sciences (2018)
