
Abstract
Schizophrenia is associated with abnormalities in the structure and functioning of white matter, but the underlying neuropathology is unclear. We hypothesized that increased tryptophan degradation in the kynurenine pathway could be associated with white matter microstructure and biochemistry, potentially contributing to white matter abnormalities in schizophrenia. To test this, fasting plasma samples were obtained from 37 schizophrenia patients and 38 healthy controls and levels of total tryptophan and its metabolite kynurenine were assessed. The ratio of kynurenine to tryptophan was used as an index of tryptophan catabolic activity in this pathway. White matter structure and function were assessed by diffusion tensor imaging (DTI) and 1H magnetic resonance spectroscopy (MRS). Tryptophan levels were significantly lower (p<0.001), and kynurenine/tryptophan ratios were correspondingly higher (p=0.018) in patients compared with controls. In patients, lower plasma tryptophan levels corresponded to lower structural integrity (DTI fractional anisotropy) (r=0.347, p=0.038). In both patients and controls, the kynurenine/tryptophan ratio was inversely correlated with frontal white matter glutamate level (r=−0.391 and −0.350 respectively, p=0.024 and 0.036). These results provide initial evidence implicating abnormal tryptophan/kynurenine pathway activity in changes to white matter integrity and white matter glutamate in schizophrenia.
Similar content being viewed by others


INTRODUCTION
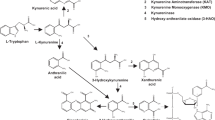
Decreased plasma concentration of tryptophan was initially identified in antipsychotic-free schizophrenia patients over 40 years ago (Manowitz et al, 1973). Several subsequent studies have also found reduced plasma tryptophan in schizophrenia patients, although findings of no significant decrease have also been reported (summarized in Table 1). Because over 90% of tryptophan is metabolized through the kynurenine pathway (Schwarcz et al, 2012) controlled by rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO), the reduction of tryptophan in schizophrenia has been attributed to an increased conversion from tryptophan to kynurenine metabolites, as supported by higher kynurenine/tryptophan (KYN/TRP) ratio (Barry et al, 2009; Schwieler et al, 2015). However, an unanswered question is how abnormal tryptophan metabolism may contribute to brain abnormalities identified in patients with schizophrenia.
IDO regulation of kynurenine pathway metabolism is thought to modulate the activation of myelin-specific T cells (Platten et al, 2005). These activated T cells have been shown to generate proinflammatory cytokines that can directly contribute to demyelination and indirectly potentiate antibodies against myelin proteins and also increase perivascular infiltrates (Vass et al, 1992; Kroenke and Segal, 2011). Animal studies have established that abnormal elevation of kynurenine in early development may result in decreased dendritic spine density and glutamatergic abnormalities in adults, as well as cognitive deficits resembling those observed in schizophrenia (Pocivavsek et al, 2012; Pershing et al, 2015). These preclinical data suggest a possible impact of abnormal kynurenine pathway metabolism on cerebral white matter structure and/or biochemistry. A previous study in psychiatrically healthy elderly individuals found that low plasma tryptophan levels were associated with more deep white matter lesions (Yao et al, 1999), further supporting a possible link between abnormal tryptophan metabolism that could be due to intake, availability, and/or degradation, and changes in the white matter.
Indeed, accumulating evidence indicates that white matter abnormality is one of the more consistent characteristics of schizophrenia pathophysiology (Kochunov and Hong, 2014; Davis et al, 2003) and likely contributes to symptoms of schizophrenia through impaired synchronization of brain networks (Bartzokis, 2002). Much of the recent evidence for white matter abnormalities comes from use of diffusion tensor imaging (DTI) that measures the directional diffusion of water. Fractional anisotropy (FA) is a commonly used metric obtained with DTI that is reduced in schizophrenia (Ellison-Wright and Bullmore, 2009) and sensitive to aging, inflammation, and demyelination (Werring et al, 1999; Song et al, 2005; Kochunov et al, 2008). One of our aims is to test the hypothesis that peripheral levels of tryptophan and kynurenine are related to white matter neurobiology in schizophrenia as indexed by cerebral white matter FA.
We also acquired proton magnetic resonance spectroscopy (1H-MRS) from a frontal white matter region to investigate a potential link between tryptophan metabolism and white matter glutamate levels. Previous MRS studies of glutamate in the white matter in schizophrenia found higher glutamate levels in acute psychosis (Ota et al, 2012) and elderly schizophrenia patients (Chang et al, 2007). In rodents, systemic administration of kynurenine significantly decreased extracellular glutamate levels in the prefrontal cortex (Wu et al, 2010; Konradsson-Geuken et al, 2010). However, to our knowledge no studies have examined potential effects of tryptophan/kynurenine on white matter glutamate in schizophrenia. Based on the preclinical data in the prefrontal cortex, we tested the hypothesis that peripheral levels of tryptophan and kynurenine are related to white matter glutamate levels in schizophrenia patients.
MATERIALS AND METHODS
Participants
Patients (n=37, age range 20–58 years; 30 with diagnosis of schizophrenia and 7 with diagnosis of schizoaffective disorder) were recruited from the outpatient clinics at the Maryland Psychiatric Research Center and the neighboring mental health clinics. Healthy controls (n=38, age range 20–61 years) were recruited through media advertisements. Demographics of the sample are reported in Table 2. Diagnoses were confirmed with the Structured Clinical Interview (SCID) for DSM-IV in all participants. Major medical and neurological illnesses, history of head injury with cognitive sequelae, and mental retardation were exclusionary. Patients and controls with substance dependence within the past 6 months or current substance abuse (except nicotine) were excluded. Except for four medication-free participants, all schizophrenia patients were on antipsychotic medications, including 8 taking clozapine, 4 taking typical antipsychotics, 17 taking atypicals, and 4 taking a combination of typical and atypical antipsychotics. In addition, 15 of the patients were on an antidepressant medication at the time of study. Controls had no current DSM-IV Axis I diagnoses and no family history of psychosis in the prior two generations. Participants gave written informed consent. This study was approved by the University of Maryland IRB.
Clinical Assessments
Overall psychiatric symptoms were assessed with the mean of the 20 item Brief Psychiatric Rating Scale (BPRS); subscales for positive psychosis symptoms and anxiety/depression symptoms were also generated in order to test specific symptom domains (Overall and Gorham, 1962). To explore how cognitive deficits might be related to altered levels of tryptophan and kynurenine metabolites, participants were tested with the Digit Symbol Coding task of the WAIS-3 (Wechsler, 1997) and the Digit Sequencing task from the Brief Assessment of Cognition in Schizophrenia (Keefe et al, 2004) to assess processing speed and working memory, respectively.
Biochemistry
Participants were instructed to fast overnight. Whole blood was collected between 0900 and 1030 h in EDTA-containing tubes (Vacutainer) that were immediately centrifuged at 2500 r.p.m. for 10 min. Plasma was then aliquoted into separate tubes and stored at −80 °C until assay. Total tryptophan (free plus protein bound) and kynurenine were measured by reverse-phase high-performance liquid chromatography, as previously described (Widner et al, 1997; Laich et al, 2002), using 3-nitro-L-tyrosine as an internal standard. Tryptophan was detected using an excitation wavelength of 266 nm and an emission wavelength of 366 nm. Kynurenine was detected by UV absorption at 360 nm. Retention times were 4.3 min for tryptophan and 2.3 min for kynurenine. Coefficients of variance for the assay were 1.7% for tryptophan and 4.2% for kynurenine.
Diffusion Tensor Imaging
All imagings were performed at the University of Maryland Center for Brain Imaging Research using a Siemens 3T TRIO MRI (Erlangen, Germany) system equipped with a 32-channel phase array head coil. The DTI data were collected using a single-shot, echo-planar, single refocusing spin-echo, T2-weighted sequence with a spatial resolution of 1.7 × 1.7 × 3.0 mm. The sequence parameters were: TE/TR=87/8000 ms, FOV=200 mm, axial slice orientation with 50 slices and no gaps, five b=0 images, and 64 isotropically distributed diffusion-weighted directions with b=700 s/mm2. These parameters maximized the contrast to noise ratio for FA measurements (Kochunov et al, 2012). A tract-based spatial statistics (TBSS) method was used for tract-based analysis of diffusion anisotropy (Smith et al, 2006). FA images were created by fitting the diffusion tensor to the motion and eddy current diffusion data. RMSDIFF (Smith et al, 2006) was used to estimate the root mean square movement distance between diffusion sensitized and b=0 images. All data passed QA control of <3 mm accumulated motion during the scan. In the next step, all FA images were globally spatially normalized to the Johns Hopkins University atlas (Wakana et al, 2004) and then nonlinearly aligned to a groupwise, minimal-deformation target (MDT) brain using the FLIRT method (Kochunov et al, 2001; Smith et al, 2006). Next, individual FA images were averaged to produce a group-average anisotropy image. This image was used to create a groupwise skeleton of white matter tracts. Finally, FA images were thresholded at FA=0.20 level to eliminate nonwhite matter voxels, and FA values were projected onto the groupwise skeleton of white matter structures. This step accounts for residual misalignment among individual white matter tracts. FA values were assigned to each point along a skeleton using the peak value found within a designated range perpendicular to the skeleton. Whole-brain tract-averaged FA was used as the primary measure for statistical analysis, based on the rationale that if there was a relationship between peripheral measures of tryptophan metabolism and white matter structure, the effect would be global rather than specific to any particular tract. However, as the MRS voxel was in frontal white matter (see below), we also conducted exploratory analyses using FA for corona radiata and genu of the corpus callosum. One patient did not complete DTI.
White Matter MRS
A spectroscopic voxel was placed in the white matter of the forceps minor area of the left hemisphere, corresponding to the left anterior corona radiata (ACR) under the left prefrontal cortex, avoiding CSF and gray matter. The prefrontal white matter MRS data were previously reported in the context of examining the relationship of myo-inositol and FA in the white matter in schizophrenia (Chiappelli et al, 2015); MRS data from all participants involved in the previous study are included here, with no new MRS data added to this analysis, although biochemical data from one additional schizophrenia patient who did not complete MRS are included here. Single-voxel PRESS localization was utilized with the following parameters: TR/TE=2000/30 ms, VOI ~3.4 cm3, NEX=256, 2048 complex points, 1.2 kHz spectral width, and total scan time ~12 min. A water reference (NEX=8) was collected and utilized for phasing and eddy current correction. A basis set was simulated using the GAVA software package (Soher et al, 2007) that was modified to yield a Lorentzian lineshape instead of its default Gaussian lineshape as previously reported (Rowland et al, 2015). This basis set was imported into LCModel, an automated curve fitting software package (Provencher, 1993) for metabolite quantification. Metabolites were corrected for the proportion of the gray matter, white matter, and cerebrospinal fluid (CSF) within the spectroscopic voxel using in-house Matlab code (Gasparovic et al, 2006). All metabolite concentrations were relative to the water reference and are reported in institutional units (IUs). The exclusion criteria for these data were: (1) SNR reported by LCModel was ⩽10; (2) FWHM reported by LCModel was ⩾0.09; and (3) metabolite fits with %SD (estimated standard deviations also called Cramer–Rao lower bounds) above >20. All spectra were of good quality with a mean FWHM of 0.045 for controls and 0.048 for patients (t=1.01, p=0.318) and a mean SNR of 27.7 for controls and 26.0 for patients (t=1.31, p=0.196). Five metabolites were consistently identified with %SD <20 and were used in statistical analyses: glutamate (mean %SD=7.3 (control), 7.4 (patient)), NAA plus NAAG (tNAA; mean %SD=2.7 (control), 3.1 (patient)), creatine plus phosphocreatine (tCr; mean %SD=2.9 (control), 3.1 (patient)), glycerophosphocholine plus phosophocholine (tCho; mean %SD=4.2 (control), 4.3 (patient)), and myo-inositol (mean %SD=5.3 (control), 5.1 (patient)).
Statistical Analyses
Group differences were examined with one-way ANOVA, t-test, and χ2, as indicated. Further ANOVA tests were performed to test the influence of smoking and body mass index (BMI) on group differences, as the groups were significantly different for these variables (see below). Correlations between variables were examined with Pearson’s correlation coefficients, except for variables that deviated from a normal distribution as determined by Kolmogorov–Smirnov tests, in which case Spearman’s rank correlation coefficient was used. Correlational analyses were performed on the entire sample first, and then repeated for patient and control groups separately, except for when both variables in the analyses were significantly different between groups. The α was set at 0.05 and exact p-values are reported. Bonferroni corrections were only applied for whole-sample analyses with white matter metabolites because of the large number of tests. All tests were two tailed.
RESULTS
Demographics
Patient and control groups were balanced for age and gender (Table 2). However, patients had significantly greater BMI than controls (p=0.01) and were more likely to smoke (p=0.003).
Group Differences
One-way ANOVAs comparing plasma levels of tryptophan, kynurenine, and KYN/TRP ratio between patients and controls found that patients had significantly lower levels of tryptophan (F(1, 73)=14.99, p<0.001; Figure 1) but not kynurenine (F(1, 73)=0.68, p=0.41); KYN/TRP ratio was higher in patients compared with controls (F(1, 73)=5.85, p=0.018; Table 2). Adding smoking status and BMI as covariates, tryptophan remained significantly lower in patients compared with controls (F(1, 70)=9.98, p=0.002). Tryptophan levels did not differ between smokers and nonsmokers (F(1, 70)=0.84, p=0.35), nor was there a smoking × diagnosis interaction (F(1, 70)=1.34, p=0.27). Females had lower levels of tryptophan than males (F(1, 71)=5.57, p=0.021) but there was no gender × diagnosis interaction (F(1, 71)=0.05, p=0.83). There was a significant inverse correlation between tryptophan and age in the combined sample (r=−0.254, p=0.028) but not separately in either group (both p>0.05). Tryptophan was not related to BMI in either controls (r=−0.270, p=0.10) or patients (r=0.143, p=0.40). Based on these results, subsequent analyses focused on testing the potential association of the significantly low tryptophan level and higher KYN/TRP ratios with brain imaging markers.

Scatterplot showing difference in plasma tryptophan levels.
White Matter Fractional Anisotropy
Whole-brain tract-averaged FA in patients was significantly reduced in patients compared with controls (t(72)=2.05, p=0.044). Plasma fasting tryptophan level was significantly correlated with whole-brain tract-averaged FA in patients (r=0.347, p=0.038, n=36; see Figure 2) but not in controls (r=0.182, p=0.27, n=38). Tryptophan level remained positively related to FA in patients after adding age, smoking status, and gender as covariates in linear regression (β=0.320, p=0.009). Tryptophan levels were also positively correlated with FA of corona radiata (r=0.330, p=0.050) and genu of corpus callosum (r=0.346, p=0.039) in patients. KYN/TRP ratio was not significantly correlated with FA in either controls or patients (all p>0.05).

Scatterplot displaying relationship of tryptophan to whole brain-averaged FA in patients.
White Matter Neurochemistry
As previously reported (Chiappelli et al, 2015), there were no significant differences between patients and controls in levels of glutamate (p=0.81) or the other metabolites including tCho, tNAA, tCr, or myo-inositol in this frontal white matter location (all p>0.14). In the combined sample of patients and controls, with Bonferroni correction for 15 (3 peripheral metabolites tryptophan, kynurenine, and KYN/TRP × 5 MRS metabolites) analyses (corrected α is p<0.0033), the only significant correlation was a negative correlation between KYN/TRP and frontal white matter glutamate (r=−0.359, p=0.002). Examination of this relationship with linear regression found that the KYN/TRP still predicted white matter glutamate after controlling for age, gender, and smoking status (β=−0.248, p=0.025). Further analysis showed that the negative relationship between KYN/TRP and white matter glutamate was found in both healthy controls (r=−0.350, p=0.036) and schizophrenia patients (r=−0.391, p=0.024; Figure 3), suggesting that higher tryptophan to kynurenine conversion was associated with lower glutamate level at this prefrontal white matter region, consistent with animal data found in the prefrontal cortex (Wu et al, 2010; Konradsson-Geuken et al, 2010).

Scatterplots showing relationship between plasma KYN/ TRP ratio (μmol/mmol) and glutamate levels in left frontal anterior corona radiata (levels reported in IUs) in (a) healthy controls and (b) schizophrenia patients.
Association with Clinical Variables
Among patients, tryptophan levels were not significantly related to chlorpromazine dose equivalent of antipsychotic medication (rho=0.206, p=0.25). KYN/TRP ratios were also not related to chlorpromazine dose equivalent (rho=−0.022, p=0.90). Tryptophan levels were not significantly different between patients not on medication at the time of study (M=57.9 (SD=11.3) μmol/l, n=4) and patients on antipsychotic medication (M=57.0 (SD=15.7), n=33; t=0.11, p=0.91); nor did unmedicated patients differ in KYN/TRP ratio (t=0.34, p=0.74). Tryptophan levels did not differ between patients taking an antidepressant and patients not taking an antidepressant medication (t(35)=0.26, p=0.79). KYN/TRP ratio also was not significantly different based on antidepressant use (t(35)=0.72, p=0.48). There were no significant differences between patients taking clozapine and those not treated with clozapine for tryptophan (t(35)=0.20, p=0.84) or KYN/TRP levels (t(35)=0.03, p=0.98). Tryptophan was not significantly associated with mean BPRS scores (r=−0.229, p=0.17) or mean BPRS psychosis subscale scores in the patients (r=−0.071, p=0.69). KYN/TRP was also not significantly associated with BPRS scores (r=0.20, p=0.23) or BPRS psychosis subscale scores (r=−.004, p=0.98). There was a nonsignificant trend for lower tryptophan to be associated with higher levels of BPRS anxiety/depression subscale scores in patients (r=−0.329, p=0.06). Tryptophan or KYN/TRP were not significantly associated with working memory or processing speed in patients (all p>0.31).
DISCUSSION
This study employed neuroimaging assessments to explore the relationship of tryptophan metabolism to markers of white matter health in schizophrenia. Collectively, our results suggest a reduced tryptophan level in schizophrenia, and that this may indicate an increased degradation as suggested by increased KYN/TRP ratio. Altered tryptophan metabolism appears to be associated with several white matter imaging markers: lower tryptophan was associated with lower fractional anisotropy of white matter, a nonspecific marker of white matter microstructure, and a higher KYN/TRP ratio was associated with lower glutamate levels in the frontal white matter.
Although the proper interpretation of FA regarding microanatomy and physiology remains disputed (Jones et al, 2013), this parameter is robust for clinical significance and reliability. Aging is associated with both reduced FA (Kochunov et al, 2008) and decreased plasma concentration of tryptophan (Capuron et al, 2011), but in this study tryptophan levels predicted FA in schizophrenia patients even after controlling for the age effect. The causes of white matter abnormalities in schizophrenia remain unclear. One of the possibilities explaining the link between tryptophan level and FA may in part be related to evidence of chronic inflammation in schizophrenia (Miller et al, 2011; Upthegrove et al, 2014). Proinflammatory cytokines, interferon-γ in particular, upregulate IDO, one of the rate-limiting enzymes involved in conversion of tryptophan to kynurenine (Schwarcz et al, 2012). Therefore, inflammatory mediators can upregulate the metabolism of tryptophan to kynurenine, or kynurenine to kynurenic acid and quinolinic acid by glial cells within the brain (Schwarcz et al, 2012). Depending on their relative concentration, these metabolites can protect against or exacerbate excitotoxicity, pathological processes that can directly damage myelin sheath and axons. Additional support to this hypothesis is that IDO and kynurenine pathway metabolites regulate the activation of myelin-specific T cells (Platten et al, 2005) that can directly contribute to demyelination and indirectly potentiate antibodies against myelin proteins (Kroenke and Segal, 2011; Vass et al, 1992). However, in the brain TDO mRNA and protein, but not IDO, is upregulated in schizophrenia (Miller et al, 2004; Miller et al, 2006). Furthermore, given that fasting tryptophan levels, but not KYN/TRP ratios, were correlated with whole-brain tract-averaged FA in patients, it is also worth considering that this relationship is related to reduced availability of tryptophan. Tryptophan levels are not only influenced by dietary intake, but also by the composition of the gut microbiota, as specific bacteria within the gut can either synthesize tryptophan de novo, or metabolize tryptophan, thus controlling its availability to the host (O'Mahony et al, 2015). In addition, evidence from preclinical studies indicates that tryptophan and tryptophan metabolites (both bacterially derived and host derived) have immunomodulatory effects (Zelante et al, 2013). Thus, although speculative, the findings reported here may be due to a chronic mild inflammatory state in schizophrenia that reduces tryptophan availability and/or promotes tryptophan catabolism to kynurenine metabolites toxic to oligodendrocytes and/or white matter (Sundaram et al, 2014).
The relationship between tryptophan levels and mood symptoms may also be mediated through stress reactivity, as stress hormones regulate TDO, another rate-limiting enzyme for tryptophan/kynurenine conversion. In previous work we have found that schizophrenia patients with poor stress tolerance exhibit a robust increase in salivary kynurenic acid in response to a mild psychological stressor (Chiappelli et al, 2014). Furthermore, these patients with poor stress tolerance exhibit a more prolonged cortisol response to the stressor that correlates with lower white matter FA (Nugent et al, 2015). Thus, enhanced stress reactivity represents another possible mechanism underlying the observed relationship between low tryptophan levels and impaired white matter structure in schizophrenia patients.
Evidence of altered activity of the kynurenine pathway in schizophrenia includes findings of increased concentration of kynurenine and kynurenic acid in brain tissue (Schwarcz et al, 2001; Miller et al, 2006; Sathyasaikumar et al, 2011) and in cerebrospinal fluid of schizophrenia patients (Nilsson et al, 2005; Linderholm et al, 2012). Kynurenine and kynurenic acid administration to the prefrontal cortex significantly decrease extracellular glutamate levels in rodents (Wu et al, 2010; Konradsson-Geuken et al, 2010). Here, we found that in both patients and controls, KYN/TRP ratios were inversely correlated with frontal white matter glutamate. We interpret this finding as indicating that higher conversion from tryptophan to kynurenine is associated with lower glutamate in the white matter, parallel to the rodent observation in prefrontal gray matter. Recent work has also uncovered evidence of axon–glial communication within white matter that appears to be based on glutamatergic signaling and may represent a mechanism for activity-dependent modulation of myelination (Kukley et al, 2007; Ziskin et al, 2007). Although the levels of white matter glutamate measured with MRS cannot be assumed to directly reflect axon–glial signaling, and cannot distinguish between extracellular vs intracellular levels, the set of findings reported here raise the possibility that kynurenine pathway activity may have a downstream influence on glutamate-dependent white matter activity.
As this was a cross-sectional study, we cannot draw causal conclusions regarding the pathophysiological mechanism that underlies the relationship between tryptophan metabolism and white matter abnormalities. We did not have data to approximate premorbid IQ of the patients in this study that could provide clues regarding the neurodevelopmental context of our findings. Most patients included in this study were on antipsychotic medications, and the potential confounding effect of these medications cannot be ruled out. However, we found that the chlorpromazine equivalent dose was not related to plasma tryptophan in the patients. This is consistent with an in vitro study that found no effect of antipsychotics on tryptophan metabolism in cultured peripheral blood mononuclear cells from schizophrenia patients (Krause et al, 2013). Although we excluded participants identified by diagnostic interview as having current substance abuse disorders, we did not routinely perform drug screens before the study, leaving open the possibility that recent illicit drug use could have altered laboratory or imaging results for some participants. Another limitation of this study was that only total tryptophan was measured, and that kynurenine was the only tryptophan metabolite measured; the KYN/TRP ratio may be only a crude index of IDO/TDO activity (Badawy, 2015). Furthermore, ∼5 to 10% of tryptophan is metabolized to serotonin (Schwarcz et al, 2012), a pathway that is not examined in the current study. Indeed, low levels of tryptophan have been associated with mood disturbances and our data revealed a trend toward low tryptophan being associated with greater anxiety and depression in schizophrenia patients. We must also consider the limitations posed by only having peripheral and not central measures of tryptophan and kynurenine levels. Tryptophan and kynurenine both cross the blood–brain barrier; the majority of kynurenine in the brain is derived from blood, especially in conditions of systemic inflammation (Kita et al, 2002; Schwarcz et al, 2012). However, peripheral levels of tryptophan and kynurenine are not necessarily informative about the activity of IDO/TDO activity in the brain, nor do they provide information on the relative balance of kynurenine versus kynurenic acid produced by glial cells. Further studies on the relationship of tryptophan metabolism and brain structure will require parallel animal studies to provide mechanistic explanations.
Although the results of previous studies examining levels of tryptophan in schizophrenia patients have been variable, the weighted average of effect size across these studies (−0.42; see Table 1) suggests that reduced tryptophan in the peripheral blood in schizophrenia is a replicable finding with modest effect size. Levels of tryptophan can be affected by food intake, exercise, stress, gut microbiota, and some medications (Badawy, 2015). We attempted to limit the impact of some of these factors by collecting blood samples in individuals who had been fasting overnight and who were instructed to avoid strenuous exercise the day before blood collection, although we did not precisely regulate intake, stress, or activity levels. Finally, the exploration of correlations among numerous imaging and peripheral markers presents a high risk for type I error and the results must therefore be considered as only initial evidence of a link between increased tryptophan metabolism and white matter impairment in schizophrenia patients.
In conclusion, this study found preliminary evidence that altered tryptophan metabolism is related to white matter integrity in schizophrenia. Although the data in this study are not sufficient to pinpoint the underlying mechanism of this relationship, it is possible that low tryptophan reflects both decreased availability and increased catabolism due to inflammation; and inflammation may leave patients vulnerable to white matter damage through neuroinflammatory, neurovascular, and neuroendocrine pathways. Further investigation of the mechanisms underlying this relationship may reveal clinically useful biomarkers and potential treatment targets for schizophrenia.
FUNDING AND DISCLOSURE
Dr Elliot Hong receives unrestricted research funding from Mitsubishi, Your Energy Systems LLC, and Pfizer. Funding for this study was received from National Institutes of Health grants R01MH085646, R01DA027680, R01MH094520, R01MH096263, T32MH067533, P50 MH103222, and U01MH108148; a NARSAD Young Investigator Award from the Brain and Behavior Foundation; a Distinguished Investigator Award from the American Foundation for Suicide Prevention (DIG 1-162-12); with additional funding from the Rocky Mountain MIRECC for Suicide Prevention, and the University of Maryland, Joint Institute for Food Safety and Applied Nutrition through the cooperative agreement FDU.001418 (sub award PI Postolache). The findings and conclusions in this study are those of the authors and do not necessarily represent the official positions of the FDA, VA, NIH or the American Foundation for Suicide Prevention. The other authors declare no conflict of interest.
References
Badawy AA (2015). Tryptophan availability for kynurenine pathway metabolism across the life span: Control mechanisms and focus on aging, exercise, diet and nutritional supplements. Neuropharmacology (e-pub ahead of print 23 November 2015).
Barry S, Clarke G, Scully P, Dinan TG (2009). Kynurenine pathway in psychosis: evidence of increased tryptophan degradation. J Psychopharmacol 23: 287–294.
Bartzokis G (2002). Schizophrenia: breakdown in the well-regulated lifelong process of brain development and maturation. Neuropsychopharmacology 27: 672–683.
Capuron L, Schroecksnadel S, Féart C, Aubert A, Higueret D, Barberger-Gateau P et al (2011). Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry 70: 175–182.
Carl GF, Brogan MP, Young BK (1992). Is plasma serine a marker for psychosis? Biol Psychiatry 31: 1130–1135.
Chang L, Friedman J, Ernst T, Zhong K, Tsopelas ND, Davis K (2007). Brain metabolite abnormalities in the white matter of elderly schizophrenic subjects: implication for glial dysfunction. Biol Psychiatry 62: 1396–1404.
Chiappelli J, Hong LE, Wijtenburg SA, Du X, Gaston F, Kochunov P et al (2015). Alterations in frontal white matter neurochemistry and microstructure in schizophrenia: implications for neuroinflammation. Transl Psychiatry 14: e548.
Chiappelli J, Pocivavsek A, Nugent KL, Notarangelo FM, Kochunov P, Rowland LM et al (2014). Stress-induced increase in kynurenic acid as a potential biomarker for patients with schizophrenia and distress intolerance. JAMA Psychiatry 71: 761–768.
Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR et al (2003). White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry 60: 443–456.
Ellison-Wright I, Bullmore E (2009). Meta-analysis of diffusion tensor imaging studies in schizophrenia. Schizophr Res 108: 3–10.
Fukushima T, Iizuka H, Yokota A, Suzuki T, Ohno C, Kono Y et al (2014). Quantitative analyses of schizophrenia-associated metabolites in serum: serum D-lactate levels are negatively correlated with gamma-glutamylcysteine in medicated schizophrenia patients. PLoS One 9: e101652.
Gasparovic C, Song T, Devier D, Bockholt HJ, Caprihan A, Mullins PG et al (2006). Use of tissue water as a concentration reference for proton spectroscopic imaging. Magn Reson Med 55: 1219–1226.
Jones DK, Knösche TR, Turner R (2013). White matter integrity, fiber count, and other fallacies: the do's and don'ts of diffusion MRI. Neuroimage 73: 239–254.
Keefe RS, Goldberg TE, Harvey PD, Gold JM, Poe MP, Coughenour L (2004). The Brief Assessment of Cognition in Schizophrenia: reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophr Res 68: 283–297.
Kim YK, Myint AM, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2009). Cytokine changes and tryptophan metabolites in medication-naïve and medication-free schizophrenic patients. Neuropsychobiology 59: 123–129.
Kita T, Morrison PF, Heyes MP, Markey SP (2002). Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J Neurochem 82: 258–268.
Kochunov P, Hong LE (2014). Neurodevelopmental and neurodegenerative models of schizophrenia: white matter at the center stage. Schizophr Bull 40: 721–728.
Kochunov P, Lancaster JL, Thompson P, Woods R, Mazziotta J, Hardies J et al (2001). Regional spatial normalization: toward an optimal target. J Comput Assist Tomogr 25: 805–816.
Kochunov P, Thompson PM, Coyle TR, Lancaster JL, Kochunov V, Royall D et al (2008). Relationship among neuroimaging indices of cerebral health during normal aging. Hum Brain Mapp 29: 36–45.
Kochunov P, Williamson DE, Lancaster J, Fox P, Cornell J, Blangero J et al (2012). Fractional anisotropy of water diffusion in cerebral white matter across the lifespan. Neurobiol Aging 33: 9–20.
Koike S, Bundo M, Iwamoto K, Suga M, Kuwabara H, Ohashi Y et al (2014). A snapshot of plasma metabolites in first-episode schizophrenia: a capillary electrophoresis time-of-flight mass spectrometry study. Transl Psychiatry 4: e379.
Konradsson-Geuken A, Wu HQ, Gash CR, Alexander KS, Campbell A, Sozeri Y et al (2010). Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience 169: 1848–1859.
Krause D, Weidinger E, Dippel C, Riedel M, Schwarz MJ, Müller N et al (2013). Impact of different antipsychotics on cytokines and tryptophan metabolites in stimulated cultures from patients with schizophrenia. Psychiatr Danub 25: 389–397.
Kroenke MA, Segal BM (2011). IL-23 modulated myelin-specific T cells induce EAE via an IFNγ driven, IL-17 independent pathway. Brain Behav Immun 25: 932–937.
Kukley M, Capetillo-Zarate E, Dietrich D (2007). Vesicular glutamate release from axons in white matter. Nat Neurosci 10: 311–320.
Laich A, Neurauter G, Widner B, Fuchs D (2002). More rapid method for simultaneous measurement of tryptophan and kynurenine by HPLC. Clin Chem 48: 579–581.
Lee M, Jayathilake K, Dai J, Meltzer HY (2011). Decreased plasma tryptophan and tryptophan/large neutral amino acid ratio in patients with neuroleptic-resistant schizophrenia: relationship to plasma cortisol concentration. Psychiatry Res 185: 328–333.
Linderholm KR, Skogh E, Olsson SK, Dahl ML, Holtze M, Engberg G et al (2012). Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr Bull 38: 426–432.
Manowitz P, Gilmour DG, Racevskis J (1973). Low plasma tryptophan levels in recently hospitalized schizophrenics. Biol Psychiatry 6: 109–118.
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011). Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70: 663–671.
Miller CL, Llenos IC, Dulay JR, Barillo MM, Yolken RH, Weis S (2004). Expression of the kynurenine pathway enzyme tryptophan 2,3-dioxygenase is increased in the frontal cortex of individuals with schizophrenia. Neurobiol Dis 15: 618–629.
Miller CL, Llenos IC, Dulay JR, Weis S (2006). Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res 1073-1074: 25–37.
Nilsson LK, Linderholm KR, Engberg G, Paulson L, Blennow K, Lindström LH et al (2005). Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr Res 80: 315–322.
Nugent KL, Chiappelli J, Sampath H, Rowland LM, Thangavelu K, Davis B et al (2015). Cortisol reactivity to stress and its association with white matter integrity in adults with schizophrenia. Psychosom Med 77: 733–742.
O'Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF (2015). Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 277: 32–48.
Ota M, Ishikawa M, Sato N, Hori H, Sasayama D, Hattori K et al (2012). Glutamatergic changes in the cerebral white matter associated with schizophrenic exacerbation. Acta Psychiatr Scand 126: 72–78.
Overall JE, Gorham DR (1962). The Brief Psychiatric Rating Scale. Psychol Rep 10: 799–812.
Pershing ML, Bortz DM, Pocivavsek A, Fredericks PJ, Jørgensen CV, Vunck SA et al (2015). Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: implications for schizophrenia. Neuropharmacology 90: 33–41.
Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM et al (2005). Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310: 850–855.
Pocivavsek A, Wu HQ, Elmer GI, Bruno JP, Schwarcz R (2012). Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur J Neurosci 35: 1605–1612.
Potkin SG, Cannon-Spoor HE, DeLisi LE, Neckers LM, Wyatt RJ (1983). Plasma phenylalanine, tyrosine, and tryptophan in schizophrenia. Arch Gen Psychiatry 40: 749–752.
Provencher SW (1993). Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30: 672–679.
Rao ML, Gross G, Strebel B, Bräunig P, Huber G, Klosterkötter J (1990). Serum amino acids, central monoamines, and hormones in drug-naive, drug-free, and neuroleptic-treated schizophrenic patients and healthy subjects. Psychiatry Res 34: 243–257.
Rowland LM, Summerfelt A, Wijtenburg SA, Du X, Chiappelli JJ, Krishna N et al (2015). Frontal glutamate and γ-aminobutyric acid levels and their associations with mismatch negativity and digit sequencing task performance in schizophrenia. JAMA Psychiatry 73: 1–10.
Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP et al (2011). Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr Bull 37: 1147–1156.
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012). Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13: 465–477.
Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC (2001). Increased cortical kynurenate content in schizophrenia. Biol Psychiatry 50: 521–530.
Schwieler L, Larsson MK, Skogh E, Kegel ME, Orhan F, Abdelmoaty S et al (2015). Increased levels of IL-6 in the cerebrospinal fluid of patients with chronic schizophrenia—significance for activation of the kynurenine pathway. J Psychiatry Neurosci 40: 126–133.
Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE et al (2006). Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 31: 1487–1505.
Soher BJ, Young K, Bernstein A, Aygula Z, Maudsley AA (2007). GAVA: spectral simulation for in vivo MRS applications. J Magn Reson 185: 291–299.
Song SK, Yoshino J, Le TQ, Lin SJ, Sun SW, Cross AH et al (2005). Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 26: 132–140.
Sundaram G, Brew BJ, Jones SP, Adams S, Lim CK, Guillemin GJ (2014). Quinolinic acid toxicity on oligodendroglial cells: relevance for multiple sclerosis and therapeutic strategies. J Neuroinflammation 11: 204.
Tortorella A, Monteleone P, Fabrazzo M, Viggiano A, De Luca L, Maj M (2001). Plasma concentrations of amino acids in chronic schizophrenics treated with clozapine. Neuropsychobiology 44: 167–171.
Upthegrove R, Manzanares-Teson N, Barnes NM (2014). Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res 155: 101–108.
van der Heijden FM, Fekkes D, Tuinier S, Sijben AE, Kahn RS, Verhoeven WM (2005). Amino acids in schizophrenia: evidence for lower tryptophan availability during treatment with atypical antipsychotics? J Neural Transm 112: 577–585.
Vass K, Heininger K, Schafer B, Linington C, Lassmann H (1992). Interferon gamma potentiates antibody-mediated demyelination in vivo. Ann Neurol 32: 198–206.
Wakana S, Jiang H, Nagae-Poetscher LM, van Zijl PC, Mori S (2004). Fiber tract-based atlas of human white matter anatomy. Radiology 230: 77–87.
Wechsler D (1997) Wechsler Adult Intelligence Scale—Third Edition (WAIS-III). Harcourt Assessment: San Antonio, TX.
Werring DJ, Clark CA, Barker GJ, Thompson AJ, Miller DH (1999). Diffusion tensor imaging of lesions and normal-appearing white matter in multiple sclerosis. Neurology 52: 1626–1632.
Widner B, Werner ER, Schennach H, Wachter H, Fuchs D (1997). Simultaneous measurement of serum tryptophan and kynurenine by HPLC. Clin Chem 43: 2424–2426.
Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R (2010). The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci 40: 204–210.
Xuan J, Pan G, Qiu Y, Yang L, Su M, Liu Y et al (2011). Metabolomic profiling to identify potential serum biomarkers for schizophrenia and risperidone action. J Proteome Res 10: 5433–5443.
Yao H, Yuzuriha T, Koga H, Fukuda K, Endo K, Matsumoto T et al (1999). Decreased plasma tryptophan associated with deep white matter lesions in elderly subjects. J Neurol Neurosurg Psychiatry 66: 100–103.
Yao JK, Dougherty GG Jr, Reddy RD, Keshavan MS, Montrose DM, Matson WR et al (2010). Altered interactions of tryptophan metabolites in first-episode neuroleptic-naive patients with schizophrenia. Mol Psychiatry 15: 938–953.
Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G et al (2013). Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39: 372–385.
Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE (2007). Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci 10: 321–330.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chiappelli, J., Postolache, T., Kochunov, P. et al. Tryptophan Metabolism and White Matter Integrity in Schizophrenia. Neuropsychopharmacol 41, 2587–2595 (2016). https://doi.org/10.1038/npp.2016.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.66
This article is cited by
-
Frontostriatal circuitry and the tryptophan kynurenine pathway in major psychiatric disorders
Psychopharmacology (2024)
-
Neuroimaging genetics approaches to identify new biomarkers for the early diagnosis of autism spectrum disorder
Molecular Psychiatry (2023)
-
The tryptophan catabolite or kynurenine pathway in schizophrenia: meta-analysis reveals dissociations between central, serum, and plasma compartments
Molecular Psychiatry (2022)
-
Psychological outcomes of COVID-19 survivors at sixth months after diagnose: the role of kynurenine pathway metabolites in depression, anxiety, and stress
Journal of Neural Transmission (2022)
-
Associations between expression of indoleamine 2, 3-dioxygenase enzyme and inflammatory cytokines in patients with first-episode drug-naive Schizophrenia
Translational Psychiatry (2021)
