
Abstract
The dorsolateral striatum mediates habit formation, which is expedited by exposure to alcohol. Across species, alcohol exposure disinhibits the DLS by dampening GABAergic transmission onto this structure’s principal medium spiny projection neurons (MSNs), providing a potential mechanistic basis for habitual alcohol drinking. However, the molecular and circuit components underlying this disinhibition remain unknown. To examine this, we used a combination of whole-cell patch-clamp recordings and optogenetics to demonstrate that ethanol potently depresses both MSN- and fast-spiking interneuron (FSI)-MSN GABAergic synaptic transmission in the DLS. Concentrating on the powerfully inhibitory FSI-MSN synapse, we further show that acute exposure of ethanol (50 mM) to striatal slices activates delta opioid receptors that reside on FSI axon terminals and negatively couple to adenylyl cyclase to induce a long-term depression of GABA release onto both direct and indirect pathway MSNs. These findings elucidate a mechanism through which ethanol may globally disinhibit the DLS.
Similar content being viewed by others


INTRODUCTION
The dorsolateral subregion of the striatum (DLS) mediates habits, such as ethanol (EtOH) consumption in late-stage alcoholism, while the dorsomedial subregion (DMS) mediates goal-directed action strategies (Balleine et al, 2009). Animals with DMS lesions only express habit response strategies, while those with lesions to the DLS exclusively use a goal-directed action strategy (Yin et al, 2004, 2005). Thus, the relative output of one striatal subregion over the other dictates the expression of its cognate action strategy. Animals previously exposed to EtOH respond habitually for EtOH at earlier time points than non-addictive substances such as food pellets or sucrose solution (Dickinson et al, 2002; Corbit et al, 2012). In addition, EtOH-drinking rats form habits for sucrose solution more rapidly than EtOH-naive animals (Corbit et al, 2012). A mechanistic understanding of how EtOH may interact with the DLS to achieve this is lacking.
The effects of acute and chronic EtOH exposure on dorsal striatal function provide clues as to how this drug mediates a possible global activation of the DLS. Blomeley et al (2011), as well as Wilcox et al (2014) demonstrated that acute EtOH exposure to striatal slices depresses inhibitory synaptic transmission onto principal medium spiny neurons (MSNs) of the DLS. This acute EtOH effect may persist in chronic drinking models as chronic EtOH consumption in rodents and non-human primates depresses GABA release onto MSNs of the DLS/putamen (Cuzon Carlson et al, 2011; Wilcox et al, 2014). Despite the mounting evidence of EtOH-induced disinhibition of DLS MSNs, the microcircuit and molecular determinants of EtOH-induced MSN disinhibition remain unknown.
MSNs receive a majority of inhibitory inputs from neighboring MSNs and parvalbumin (PV)-containing fast-spiking interneurons (FSIs; Koos et al, 2004; Tepper et al, 2004). MSNs synapse upon the distal dendrites of other MSNs while FSIs form multiple synapses upon MSNs perisomatically to powerfully regulate MSN output (Koos et al, 2004). The FSI population increases in density laterally through the dorsal striatum (Schlösser et al, 1999; Luk and Sadikot, 2001), positioning these cells to influence DLS output to a greater extent than DMS output. FSIs and other striatal cell types, like MSNs, express a host of presynaptically localized Gi/o-coupled receptors such as the cannabinoid type 1 receptor (CB1) and 5-HT1b, among others (Mathur et al, 2011, 2013; Atwood et al, 2014a, b). When activated, these receptors dampen neurotransmitter release. As such, EtOH may be acting through one of these receptors to dampen GABA release onto MSNs. To examine this possible mechanism of action of EtOH on the depression of inhibitory synapses on DLS MSNs, we use whole-cell patch-clamp electrophysiology and optogenetics to isolate potential EtOH effects at either MSN–MSN or FSI-MSN synapses. We show that EtOH induces LTD at MSN and FSI inputs onto MSNs, and that the powerfully inhibitory FSI-MSN synapse is depressed through activation of a presynaptic delta opioid receptor (DOR). Further, we identify adenylyl cyclase/cAMP as the signaling pathway responsible for this novel form of EtOH-induced synaptic plasticity.
METHODS
All experiments were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory and were approved by the Institutional Animal Care and Use Committee at the University of Maryland, Baltimore and the National Institute on Alcohol Abuse and Alcoholism. Mice were housed with littermates (2–5 per cage) under a 12-h light/dark cycle (lights on at 0700 hours, off at 1900 hours) with ad libitum access to food and water.
Viral-Mediated Channelrhodopsin (ChR2) Expression
To virally express ChR2 selectively in PV-containing FSIs or MSNs in the DLS, >2-month old Pvalb-cre or RGS9-cre (C57BL/6 background) transgenic mice, respectively, were stereotaxically injected with an AAV vector under an EF1a promoter containing a DIO-ChR2-mCherry or eYFP construct flanked by loxP sites (AAV-flox-DIO-ChR2-mCherry/eYFP) at a volume of 400 nl and a rate of 20 nl/min into the DLS (AP +0.6 mm, ML±2.25 mm, DV −2.4 mm from bregma), following our previously described methods (Mathur et al, 2013). To target FSIs for ChR2 expression and record from direct (MSND) or putative indirect (MSNID) MSNs, Pvalb-cre × Drd1a-tdTomato mice were injected with the same viral construct. Animals recovered for a minimum of 3 weeks before brains were sliced for electrophysiology experiments.
Brain Slice Preparation
At least 3 weeks post surgery, animals were anesthetized with isoflurane and brains were dissected out and submerged in 95% oxygen, 5% carbon dioxide (carbogen)-bubbled ice cold cutting solution (194 mM sucrose, 30 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 26 mM NaHCO3, 1.2 mM NaH2PO4, and 10 mM D-glucose). Brains were sliced coronally at 250 μm with a vibratome (Leica VT 1200) and stored in carbogen-bubbled artificial cerebrospinal fluid (aCSF; 124 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 26 mM NaHCO3, 1.2 mM NaH2PO4, and 10 mM D-glucose). Sections were incubated at 32.4 °C for 30 min before they were removed and stored at room temperature until electrophysiological recordings were performed. Before recording, slices were hemisected and were constantly perfused with carbogen-bubbled aCSF (29–31 °C) via a gravity perfusion system throughout the experiment.
Whole-Cell Voltage-Clamp Recordings
Injection sites of ChR2-mCherry/eYFP into the DLS were visualized through the epifluorescent light path using a mercury bulb lamp (X-Cite series 120Q). Whole-cell recordings were made from DLS MSNs receiving inputs from ChR2 infected cells as described previously (Mathur et al, 2013). Optically evoked IPSCs (oIPSC) were recorded using borosilicate glass pipettes with resistances in the 2–5 MΩ range. Pipettes were pulled on a Flaming Brown micropipette puller (Sutter Instruments) and filled with a CsCl-based internal solution (150 mM CsCl, 10 mM HEPES, 2 mM MgCl2, 0.3 mM Na-GTP, 5 mM QX-314, 3 mM Mg-ATP, and 0.2 mM BAPTA) with osmolarity ranging from 305 to 310 mOsm at a pH of 7.3. oIPSCs were evoked with blue light (473 nm) using epifluorescent LED field illumination (Lumen Dynamics, XLED1). Blue light-evoked test pulses were delivered every 20 s with a pulse duration of 2–4 ms. Blue light intensity was adjusted to record oIPSCs with an average amplitude of 600 pA. Ultraviolet light (370 nm, pulse width of 4 ms) was used to photo-uncage bath-applied caged GABA (CNB-GABA, 50 μM) to measure uncaged IPSCs (uIPSC). Electrically evoked IPSCs (eIPSC) were induced using a concentric bipolar stimulating electrode (World Precision Instruments) located locally in the DLS approximately 100 μm from the recorded neuron. eIPSCs were recorded with AP5 (50 μM) and NBQX (5 μM) present in the bath. MSNs were voltage clamped at −60 mV using a MultiClamp 700B amplifier (Molecular Devices) and recordings were filtered at 2 kHz and digitized at 10 kHz. Clampex 10.4.1.4 software (Molecular Devices) was used for data acquisition. Cells were discarded from analysis if series resistance changed by more than 15% throughout the course of the experiment.
Statistical Analysis
Mean IPSC amplitudes were measured in Clampfit 10.4.1.4 and analyzed using GraphPad Prism 6.01. IPSC amplitudes were averaged per minute and expressed as a percent change from baseline measurements. Average baseline IPSC amplitudes and amplitudes in the final 5 min of recording were compared using a two-tailed paired t test. Changes in IPSC amplitudes between experiments were analyzed using a two-tailed unpaired t test.
Drugs
EtOH, SrCl2 and GDP-β-S were purchased from Sigma-Aldrich. Cell-impermeant BAPTA tetracesium salt (molecular weight of 1004.03) and CNB-GABA were purchased from Invitrogen. All other drugs were purchased from Tocris Bioscience.
RESULTS
EtOH Depresses MSN- and FSI-MSN Synapses
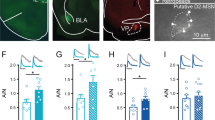
The effect of EtOH on MSN- and FSI-MSN synaptic transmission was tested by measuring oIPSC amplitudes during and after 10 min acute exposure to 50 mM EtOH, a concentration of EtOH that correlates to a blood alcohol concentration (BAC) of 230 mg/dl in humans. This BAC is consistent with what late-stage alcoholics achieve (Brick and Erickson, 2009). EtOH depressed oIPSC amplitude of MSN inputs onto MSNs (MSN–MSN) to 75.69±7.03% of baseline (t=4.06, df=4, p=0.02, Figure 1a) in 5 out of 5 cells recorded. FSI inputs onto MSNs were depressed to 70.79±5.733% of baseline following EtOH application (t=5.1, df=18, p<0.001, Figure 1b). EtOH-induced depression at this synapse was not due to decreased sensitivity of ChR2 to blue light during the 30 min experiment, as the amplitude of FSI-MSN oIPSCs was stable across 30 min of recording in the absence of EtOH (Figure 1b filled circles; 91.75±9.76% of baseline, t=0.88, df=9, p=0.42). Further, EtOH-induced depression of the FSI-MSN synapse was significantly different from the no drug control (t=2.9, df=27, p=0.01). Unlike the MSN–MSN synapse, EtOH depressed FSI-MSN transmission in 14 out of the 19 cells recorded. EtOH-induced depression of both MSN- and FSI-MSN synapses persisted even 15 min following the end of EtOH application. Thus, we call this depression EtOH-LTD. Importantly, neither MSN–MSN nor FSI-MSN EtOH-LTD was due to a change in postsynaptic neuron series resistance (Figure 1c and d). We next determined if a range of EtOH concentrations that are achievable in vivo are capable of producing EtOH-LTD at the FSI-MSN synapse. Both 10 and 80 mM EtOH (the corresponding BAC values of which are 46 and 368 mg/dl, respectively) depressed the FSI-MSN synapse (Figure 1e). Similar to 50 mM, 10 mM EtOH depressed FSI transmission onto MSNs to 73.72±10.96% of baseline (t=2.4, df=8, p=0.04), while 80 mM EtOH depressed FSI-MSN transmission to 79.93±7.20% of baseline (t=2.79, df=8, p=0.02). To determine if the heterogeneity of the EtOH response at the FSI-MSN synapse is due to direct pathway MSN (MSND) or indirect pathway MSN (MSNID) DLS output specificity, we crossed Pvalb-cre mice with bacterial artificial chromosome-transgenic mice expressing tdTomato under the D1-dopamine receptor promoter. We recorded the effect of EtOH at FSI- D1-containing (tdTomato positive) MSNsD or FSI-putative D2-expressing MSNsID (tdTomato negative). EtOH depressed FSI transmission onto both MSNsD and MSNsID; MSND oIPSC amplitude decreased to 79.83±8.76% of baseline, t=2.30, df=12, p=0.04; MSNID oIPSC amplitude was 74.74±6.69% of baseline, t=3.78, df=13, p=0.002 (Figure 1f), indicating EtOH-LTD is not pathway specific. The variation in the timing of onset of EtOH-LTD that occurs between FSI-MSND and FSI-MSNsID could occur due to the percentage of cells in each group that respond to the EtOH application. Alternatively, this difference may be a function of the Pvalb-cre × Drd1a-tdTomato genotype. Because FSIs are the sole source of perisomatic synapses on MSNs (Koos et al, 2004; Tepper et al, 2004), they provide the most powerful inhibitory influence of all sources of GABA onto MSNs and, thus, are positioned to play a large role in EtOH effects on the disinhibition of the DLS. As such, we henceforth concentrated on elucidating the mechanism of EtOH-LTD at the FSI-MSN synapse.

Ethanol (EtOH) depresses medium spiny neuron (MSN) and fast-spiking interneuron (FSI) transmission onto MSNs. (a) EtOH application (10 min) depresses MSN–MSN optically evoked inhibitory postsynaptic current (oIPSC) amplitude (n=5 cells, p<0.05). (b) EtOH depresses FSI inputs onto MSNs (open circles, n=19 cells, p<0.05). Changes in FSI-MSN strength are not owing to channelrhodopsin rundown, as a no drug control does not change synaptic strength (filled circles, n=10, p>0.05). (c) Top, raw data demonstrating that EtOH-induced depression of the MSN–MSN synapse is not due to changes in series resistance (Rs, bottom). (d) Top, raw data demonstrating that EtOH-induced depression of the FSI-MSN synapse is not caused by a change in Rs, bottom. (e) 10 mM (open circles) and 80 mM (filled circles) EtOH depress FSI-MSN synaptic strength (10 mM: n=9, p<0.05; 80 mM: n=9, p<0.05). (f) EtOH depresses FSI-direct pathway MSN (MSND) synapses (filled circles, n=13 cells, p<0.05) and FSI-indirect pathway MSN (MSNID) synapses (open circles, n=14 cells, p<0.05). All scale bars represent 300 pA (vertical) and 100 ms (horizontal). All example traces depict baseline (dark) and last 5 min of the experiment (light). Schematics represent optogenetic activation of MSNs or FSIs and whole-cell patch-clamp recordings from MSNs. All error bars indicate SEM.
FSI-MSN EtOH-LTD is Presynaptically Expressed
LTD of inhibitory synapses onto MSNs that is induced at a hyperpolarized or ‘downstate’ membrane voltage of −80 mV (iLTD) is present at FSI-MSN synapses and occurs through activation of presynaptically localized CB1 (Mathur et al, 2013). To test if EtOH-LTD at the FSI-MSN synapse is presynaptically expressed and, therefore, mutually occlusive with eCB-LTD, low frequency electrical stimulation (LFS, 1 Hz, 80 pulses) was locally delivered while voltage clamping MSNs at −80 mV in the presence or absence of EtOH. LFS depressed the FSI-MSN synapse, consistent with our previous findings (Mathur et al, 2013; oIPSC amplitude 79.17±6.94% of baseline, t=3.00, df=5, p=0.03). Application of 50 mM EtOH following iLTD induction did not further depress the synapse (iLTD amplitude=79.17±6.94% of baseline compared with EtOH-LTD amplitude=80.38±9.49% of baseline, t=0.42, df=5, p=0.69, Figure 2a). Additionally, induction of iLTD following the generation of EtOH-LTD did not further depress the synapse (EtOH-LTD amplitude=89.02±5.73% of baseline compared with iLTD amplitude=83.70±6.82%, t=1.08, df=5, p=0.33, Figure 2b).

FSI-MSN EtOH-induced long-term depression (EtOH-LTD) is presynaptically expressed. (a) EtOH-LTD is mutually occlusive with endocannabinoid-LTD. Delivering electrical low frequency stimulation while voltage clamping MSNs at −80 mV (LFS, arrow) results in a depression of the FSI-MSN synapse that is not further depressed by EtOH (last 15 min, n=6 cells, p>0.05). (b) EtOH application depresses FSI-MSN synapses and is not further depressed by electrical LFS (arrow, n=6 cells, p>0.05). (c) Left panel, optically evoked, Sr2+ enabled IPSC (osIPSC) inter-event interval (IEI) increases after EtOH application (filled circles) compared with baseline (open circles, n=9 cells, **p<0.01). osIPSC amplitude does not change following EtOH application (filled circles) compared with baseline amplitudes (open circles, n=8 cells, p<0.05). Right panel, representative traces of osIPSC events during baseline (open circle) and EtOH application (filled circle). Black circles indicate osIPSC events. (d) Including 20 mM BAPTA in the internal pipette solution does not eliminate FSI-MSN EtOH-LTD (n=13 cells, p<0.05). (e) EtOH depresses electrically evoked IPSCs (eIPSC, open circles, n=5 cells, p<0.05) but does not depress CNB-GABA photo-uncaged IPSCs (uIPSC, filled circles, n=6 cells, p>0.05). (f) Inclusion of GDP-β-S into the internal pipette solution does not eliminate EtOH-LTD (n=6 cells, p<0.05). Scale bars represent 300 pA (vertical) and 100 ms (horizontal). Example traces depict baseline (dark) and the last 5 min of the experiment (light). All error bars represent SEM.
To further assess whether FSI-MSN EtOH-LTD is expressed pre- or postsynaptically, we replaced CaCl2 in the aCSF with 2 mM SrCl2 to enable circuit-specific-asynchronous transmitter release. Changes in optically elicited Sr2+-enabled asynchronous IPSC (osIPSC) event frequency and amplitude were measured in the presence of EtOH (Choi and Lovinger, 1997). EtOH increased inter-event interval (IEI; 149.7±13.06 ms in EtOH compared with control: 110.2±7.47 ms, t=4.11, df=8, p=0.003) but had no effect on event amplitude (EtOH: 23.93±6.0 pA compared with control: 27.27±7.7 pA, t=1.26, df=7, p=0.25, Figure 2c), further suggesting EtOH-LTD is expressed presynaptically. We next tested if EtOH-LTD is caused by postsynaptic Ca2+-mediated processes that could result in either GABAA endocytosis or the production of a retrograde transmitter (Della Rocca et al, 1999; Gerdeman et al, 2002) by loading the postsynaptic cell with cell-impermeable BAPTA (20 mM). BAPTA loading did not block EtOH-LTD (83.16±6.3% of baseline, t=2.67, df=12, p=0.02, Figure 2d). To isolate potential changes on the postsynaptic side, we elicited IPSCs by photo-uncaging the caged-GABA compound CNB-GABA (50 μM). Photo- uIPSC amplitudes during EtOH application did not depress in the presence of EtOH (uIPSC amplitude=103.6±3.91%, t=0.93, df=5, p=0.40), unlike eIPSC amplitudes in the same cell (eIPSC amplitude=70.47±6.21%, t=4.76, df=5, p=0.01, Figure 2e), further suggesting EtOH-LTD is presynaptically expressed. Electrically evoked IPSC amplitude depression following EtOH application was significantly different from uIPSC amplitude (uIPSC amplitude 103.6±3.91% compared with eIPSC amplitude 70.47±6.21%, t=4.52, df=10, p=0.001). Finally, to test if contributions from postsynaptic G-protein-coupled receptors underlie FSI-MSN EtOH-LTD, the G-protein receptor inhibitor GDP-β-S (500 μM) was included in the internal pipette solution. EtOH application in the presence of GDP-β-S depressed the synapse (oIPSC amplitude=71.20±6.67% of baseline, t=4.32, df=5, p=0.01, Figure 2f), indicating that contributions of postsynaptic G-protein-coupled receptors are not necessary for EtOH-LTD.
FSI-MSN EtOH-LTD is DOR Dependent
Our data show that EtOH-LTD is mutually occlusive with iLTD in the DLS, which is dependent on activation of the presynaptically localized, Gi/o-coupled CB1 (Mathur et al, 2013). To investigate if EtOH-LTD acts through this same CB1-dependent pathway, we pre-incubated slices in the CB1 receptor antagonist AM251 (5 μM) for at least 35 min before exposing EtOH to the slice. EtOH application in the presence of AM251 depressed both MSN and FSI transmission onto MSNs (MSN–MSN: oIPSC amplitude=71.31±7.31% of baseline, t=3.81, df=4, p=0.02; FSI-MSN: oIPSC amplitude=66.90±7.46% of baseline, t=4.44, df=3, p=0.021), indicating EtOH-LTD is CB1 independent.
Like CB1-dependent LTD, several other forms of LTD are known to depend on activation of a presynaptically localized Gi/o-coupled receptor (Atwood et al, 2014b). Given the known interactions between EtOH and the opioid system (Volpicelli et al, 1992; Drews and Zimmer, 2010), and that activation of these Gi/o-coupled opioid receptors is capable of inducing striatal presynaptic LTD (Atwood et al, 2014a), we next assessed whether EtOH-LTD occurs in an opioid receptor-dependent fashion. MSN–MSN EtOH-LTD was not eliminated in the presence of naloxone, a pan opioid receptor antagonist (oIPSC amplitude=80.03±6.94% of baseline, t=2.88, df=6, p=0.03, Figure 3a). However, FSI-MSN EtOH-LTD was eliminated in the presence of naloxone (5 μM, oIPSC amplitude=97.87±2.99%, t=0.71, df=6, p=0.50, Figure 3b) and was significantly different from control EtOH-induced depression of oIPSC amplitude (EtOH-LTD oIPSC amplitude=67.51±7.78% compared with naloxone oIPSC amplitude 97.87±2.99%, t=3.28, df=14, p=0.01), indicating EtOH-LTD at the MSN- and FSI-MSN synapses are mechanistically distinct.

EtOH-LTD is delta opioid receptor (DOR) dependent. (a) The pan opioid receptor antagonist, naloxone, does not eliminate EtOH-LTD at the MSN–MSN synapse (n=7 cells, p<0.05). (b) Naloxone eliminates EtOH-LTD at the FSI-MSN synapse (filled circles, n=7 cells, p>0.05), compared with control artificial cerebrospinal fluid (aCSF) (open circles, n=9 cells, p<0.05). (c) Blocking DOR with the antagonist naltrindole eliminates EtOH-LTD at the FSI-MSN synapse (filled circles, n=8 cells, p>0.05), compared with controls (open circles, n=8 cells, p<0.05). (d) EtOH-LTD is stable in the presence of naltrindole 15 min following EtOH washout (n=6, p<0.05). (e) The DOR agonist, DPDPE depresses the FSI-MSN synapse (open circles, n=12 cells, p<0.05), but the mu opioid receptor agonist, DAMGO, does not (filled circles, n=6 cells, p>0.05). (f) DPDPE depresses both FSI-MSND (filled circles, n=9 cells, p<0.05) and FSI-MSNID synapses (open circles, n=7 cells, p<0.05). (g) Incubating slices in the adenylyl cyclase activator forskolin eliminates DPDPE-induced LTD of the FSI-MSN synapse (filled circles, n=8 cells, p>0.05), compared with control aCSF (open circles, n=8 cells, p<0.05). (h) Incubating slices in the cAMP analog 8-bromo-cAMP also eliminates the DPDPE-induced LTD (filled circles, n=11 cells, p>0.05) compared with control aCSF (open circles, n=8 cells, p<0.05). All scale bars indicate 300 pA (vertical) and 100 ms (horizontal). Example traces represent baseline (dark) and the last 5 min of the experiment (light). Schematics depict optogenetic activation of MSNs or FSIs and whole-cell patch-clamp recordings from MSNs. All error bars indicate SEM.
To determine the specific opioid receptor involved in FSI-MSN EtOH-LTD, we then incubated slices in the DOR-specific antagonist naltrindole (1 μM) before applying EtOH. Naltrindole blocked EtOH-LTD at the FSI-MSN synapse (oIPSC amplitude=97.65±7.32% of baseline, t =0.32, df=7, p=0.76, Figure 3c), and the EtOH effect in this condition differed from control EtOH-LTD (control EtOH-LTD oIPSC amplitude 66.67±6.54%, t=3.16, df=14, p=0.01). To determine if this form of LTD is labile or static (Atwood et al, 2014b), we applied naltrindole (1 μM) to slices 15 min after EtOH washout (Figure 3d). EtOH-LTD did not return to baseline in the presence of naltrindole, suggesting this form of LTD is static (oIPSC following EtOH application=75.95±3.07% of baseline, t=6.23, df=5, p=0.02; oIPSC in naltrindole=63.58±9.91% of baseline, t=3.58, df=5, p=0.02). Because EtOH-LTD appears to be presynaptically expressed, and DOR, like CB1, may reside presynaptically, we hypothesized that DOR resides on the FSI terminal. Supporting this idea, application of the DOR agonist DPDPE (500 nM) depressed the FSI-MSN synapse (oIPSC amplitude=81.98±3.49% of baseline, t=5.16, df=11, p=0.0003) but the mu opioid receptor agonist, DAMGO (2 μM) did not (oIPSC amplitude=99.50±7.50% of baseline, t=0.12, df=5, p=0.91, Figure 3e). Using PV-cre × Drd1a-tdTomato mice we determined that DPDPE-induced LTD of oIPSC amplitude at the FSI-MSN synapse is not pathway specific; DPDPE depressed the FSI-MSND synapse to 66.46±8.36% of baseline (t=4.01, df=8, p=0.004) and depressed the FSI-MSNID synapse to 79.19±5.45% of baseline (t=3.82, df=6, p=0.01, Figure 3f). These data suggest that DOR is present on FSI terminals impinging upon both MSND and MSNID.
DOR-Mediated Depression of the FSI-MSN Synapse is Adenylyl Cyclase Dependent
DOR is a Gi/o-coupled receptor that is known to decrease cAMP production when activated (Al-Hasani and Bruchas, 2011). Therefore, we tested the prediction that activating adenylyl cyclase, the enzyme responsible for catalyzing the conversion of ATP to cAMP, will block DOR-induced LTD. Pre-incubating slices in 15 μM forskolin, an activator of adenylyl cyclase, for at least 20 min before DPDPE application indeed blocked DOR-induced LTD of oIPSC amplitude (oIPSC amplitude 97.04±5.10% of baseline, t=0.58, df=7, p=0.58, Figure 3g) as compared with control aCSF (oIPSC amplitude 67.04±10.65% of baseline, t=3.10, df=7, p=0.02). To test the downstream target of adenylyl cyclase, we incubated slices in the cell-permeable cAMP analog, 8-bromo-cAMP (160 μM) and measured oIPSC amplitude following DPDPE application. We found that DPDPE did not decrease oIPSC amplitude in the presence of 8-bromo-cAMP (95.77±9.67%, t=0.44, df=10, p=0.67, Figure 3h), as it did in control aCSF (68.67±5.51%, t=5.68, df=7, p=0.001), which suggests that DPDPE-induced LTD occurs through a cAMP-dependent signaling pathway. 8-bromo-cAMP blockade of DPDPE-induced LTD was significantly different than control LTD (t=2.20, df=17, p=0.04).
DOR is Located Presynaptically on FSIs
DOR is known to co-localize with a subset of GABAergic neurons in mouse dorsal striatum (Scherrer et al, 2006), a finding that suggests co-localization onto a GABAergic interneuron population as opposed to the principal GABAergic MSN. These findings are consistent with our results that EtOH-LTD is presynaptically expressed and mediated through DOR activation. Thus, we tested the prediction that DOR is located on presynaptic FSI terminals by measuring changes in asynchronous osIPSC amplitude and event frequency in the presence of DPDPE. DPDPE application increased the IEI of osIPSC events (baseline IEI 95.09±2.81 ms; post-DPDPE IEI 130.1±7.60 ms, t=4.35, df=15, p=0.001, Figure 4a). DPDPE application also depressed osIPSC amplitude (baseline amplitude 20.61±1.50 pA; post-DPDPE amplitude 16.47±1.90 pA, t=3.65, df=14, p=0.003, Figure 4a). These data suggest that DPDPE lowers GABA release probability from FSIs. In addition, there is either a possible postsynaptic effect of DPDPE on GABAA receptor availability or the reduction in probability of release reduces synchronous spontaneous event probability, which would in turn reduce event amplitude. To rule out contributions of postsynaptic DOR to FSI-MSN DPDPE-induced LTD, GDP-β-S (500 μM) was included in the internal pipette solution to disrupt G protein signaling in the postsynaptic cell. DPDPE application reliably depressed oIPSC amplitudes with GDP-β-S present (oIPSC amplitude=70.08±10.75% of baseline, t=2.78, df=8, p=0.02, Figure 4b). Finally, to test if DPDPE-induced LTD results in the generation and liberation of a Ca2+-induced retrograde signaling molecule that could signal presynaptically, we chelated postsynaptic Ca2+ using a high-BAPTA internal pipette solution (20 mM) and measured changes in oIPSC amplitude following DPDPE application. BAPTA (20 mM) in the postsynaptic cell did not block the DPDPE-induced LTD (82.37±7.27% of baseline, last 5 min, t=2.43, df=10, p=0.04, Figure 4c), suggesting postsynaptic Ca2+ is not necessary for this LTD.

Delta opioid receptors are located on FSI terminals. (a) Left panel, DPDPE increases IEI of osIPSCs and decreases the amplitude of osIPSC events (filled circles) compared with baseline (open circles, n=16 cells, ***p<0.001, **p<0.01). Right panel, representative traces of osIPSC events before DPDPE application (open circle) and after (filled circle). Black circles indicate detectable events. (b) The G-protein-coupled receptor blocker GDP-β-S does not eliminate DPDPE-induced LTD at the FSI-MSN synapse (n=9 cells, p<0.05). (c) Postsynaptic calcium chelation with BAPTA does not eliminate DPDPE-induced LTD at the FSI-MSN synapse (n=11 cells, p<0.05). Scale bars represent 300 pA (vertical) and 100 ms (horizontal). All example traces depict baseline (dark) and the last 5 min of the experiment (light). All error bars represent SEM. osIPSC, optically elicited Sr2+-enabled asynchronous IPSC.
DISCUSSION
The data presented here describe a novel, EtOH-induced, DOR-dependent LTD of FSI-MSN synaptic transmission that, in conjunction with EtOH-LTD at MSN–MSN synapses, likely contributes to the increase in DLS output that occurs following EtOH exposure. We show that DOR-induced LTD at the FSI-MSN synapse occurs through the canonical adenylyl cyclase/cAMP pathway downstream of DOR activation (Figure 5). The discovery that DOR is positioned to regulate FSI-MSN synaptic transmission implicates the opioid system in the physiological expression of habit learning (Wassum et al, 2009) and other striatal-dependent behaviors. Finally, EtOH also reliably depresses the MSN–MSN synapse. Although additional work is necessary to unveil the mechanism underlying this effect, our data thus far suggest a presynaptic locus of expression of this form of LTD that functions independently of both CB1 and opioid receptors.

Proposed mechanism of EtOH-LTD. EtOH depresses FSI-MSND and -MSNID transmission by activating DOR on presynaptic FSI terminals. DOR activation decreases adenylyl cyclase (AC) activity, which in turn decreases cAMP levels. This form of DOR-mediated synaptic plasticity occludes CB1 receptor-mediated LTD (gray receptor, grayed out inhibition of AC). FSIs (circles, right) are more prevalent in the dorsolateral striatum (DLS) than the dorsomedial striatum. EtOH-LTD of FSI-MSN synapses is thus positioned to promote a global disinhibition of the dorsolateral striatum. EtOH, ethanol; FSI, fast-spiking interneuron; MSN, principal medium spiny neurons. A full color version of this figure is available at the Neuropsychopharmacology journal online.
We demonstrate that EtOH-LTD is mutually occlusive with a form of eCB-LTD at the FSI-MSN synapse but is not mediated through CB1 receptors (Figure 5). These findings are in line with previous work demonstrating that EtOH blocks eCB-mediated disinhibition of the DLS in a CB1-independent manner (Adermark and Lovinger 2009; Clarke and Adermark, 2010). This full blockade of eCB-mediated DLS disinhibition by EtOH also suggests that the mechanism of action of EtOH-LTD at the MSN–MSN synapse is activation of a presynaptically localized Gi/o-coupled receptor capable of occluding CB1 activation-induced depression of GABA release. Given that the present findings that EtOH-LTD occludes CB1-mediated LTD, this suggests that both MSN–MSN and FSI-MSN synapses are normally under manifold control by multiple presynaptically localized Gi/o-coupled receptors. In the presence of EtOH, however, the DOR-mediated pathway overrides physiological opioid and eCB control of this synapse, possibly contributing to acute intoxication and/or lasting disinhibition of the DLS over time. Yet other presynaptic Gi/o-coupled receptors, such as the kappa opioid receptor may also reside on the FSI terminal and the possible occlusion of their effects by EtOH will also have to be examined in future studies to gain a complete picture of the disruption of physiological control over FSI-MSN synapses by EtOH.
The mechanisms through which EtOH induces release of an endogenous DOR agonist and the cellular source of this release remain unknown. A potential candidate for the source is MSNsID; opioids are known to act as Ca2+-dependent retrograde signaling molecules (Iremonger and Bains, 2009; Wamsteeker Cusulin et al, 2013) and MSNsID contain pre-proenkephalin, the precursor for the most efficacious endogenous agonist of DOR (Gerfen et al, 1990; Surmeier et al, 1996). However, we found that EtOH depresses FSI transmission onto both MSNsD and MSNsID and this depression is not eliminated with a postsynaptic Ca2+ chelator. Therefore, it is unlikely that EtOH induces release of a retrograde opioid signal from postsynaptic MSNs. It is possible that another source providing the endogenous DOR agonist is recruited by EtOH. One candidate is the external segment of the globus pallidus. This structure sends GABAergic projections to both MSNs and FSIs in the dorsal striatum (Bevan et al, 1998; Mallet et al, 2012; Mastro et al, 2014) and these projections express pre-proenkephalin (Mallet et al, 2012). Given that the variability in the induction of EtOH-LTD at the FSI-MSN synapse is not explained by differences in FSI-MSND and FSI-MSNID pathways, and that DPDPE application consistently induces FSI-MSN synaptic depression, the heterogeneous expression of FSI-MSN EtOH-LTD may be due to the variability in the EtOH-induced release of the endogenous DOR agonist.
While our results specifically describe the effects of EtOH on GABAergic transmission in the DLS, Wilcox et al (2014) report that acute EtOH exposure and chronic EtOH drinking induces a higher frequency of basal miniature IPSC (mIPSC) events in the DMS, and a lower frequency of mIPSC events in the DLS. This DMS to DLS response difference may be explained by the higher expression of FSIs in the DLS (Schlösser et al, 1999; Luk and Sadikot, 2001), as the majority of mIPSCs onto MSNs arise from FSIs (Koos et al, 2004). Taking the DOR-mediated depression of FSI-MSN synapses in the DLS following acute EtOH exposure together with the reported increase in GABA transmission onto DMS MSNs, EtOH appears to shape the global output of the striatum by modulating GABA synapses. Indeed, the importance of the presently described mechanism in drinking is supported by the findings of Nielsen et al (2012) that show striatal DOR blockade decreases repeated EtOH intake in rats, while activating striatal DOR increases consumption. In light of this, additional studies are necessary to understand the effects of EtOH on DOR-mediated depression of the FSI-MSN synapse in the DLS following a chronic drinking paradigm.
Funding and Disclosure
For this work, MHP and BMR were supported by the National Institute on Alcohol Abuse and Alcoholism of the US National Institutes of Health (grant K22 AA021414 to BNM), the Division of Intramural Clinical and Biological Research of the NIAAA/NIH (DML) and the Integrative Neuroscience Initiative on Alcoholism of the NIAAA/NIH (DML). The authors declare no conflict of interest.
References
Adermark L, Lovinger DM (2009). Frequency-dependent inversion of net striatal output by endocannabinoid-dependent plasticity at different synaptic inputs. J Neurosci 29: 1375–1380.
Al-Hasani R, Bruchas MR (2011). Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115: 1363–1381.
Atwood BK, Kupferschmidt DA, Lovinger DM (2014a). Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nature Neurosci 17: 540–548.
Atwood BK, Lovinger DM, Mathur BN (2014b). Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends in Neurosci 37: 663–673.
Balleine BW, Liljeholm M, Ostlund SB (2009). The integrative function of the basal ganglia in instrumental conditioning. Behav Brain Res 199: 43–52.
Bevan MD, Booth PA, Eaton SA, Bolam JP (1998). Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci 18: 9438–9452.
Blomeley CP, Cains S, Smith R, Bracci E (2011). Ethanol affects striatal interneurons directly and projection neurons through a reduction in cholinergic tone. Neuropsychopharmacology 36: 1033–1046.
Brick J, Erickson CK (2009). Intoxication is not always visible: an unrecognized prevention challenge. Alcohol Clin Exp Res 9: 1489–1507.
Choi S, Lovinger DM (1997). Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci 17: 8613–8620.
Clarke RB, Adermark L (2010). Acute ethanol treatment prevents endocannabinoid-mediated long-lasting disinhibition of striatal output. Neuropharmacology 58: 799–805.
Corbit LH, Nie H, Janak PH (2012). Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry 72: 389–395.
Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR et al (2011). Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology 36: 2513–2528.
Della Rocca GJ, Mukhin YV, Garnovskaya MN, Daaka Y, Clark GJ, Luttrell LM et al (1999). Serotonin 5-HT1A receptor-mediated Erk activation requires calcium/calmodulin-dependent receptor endocytosis. Biol Chem 274: 4749–4753.
Dickinson A, Wood N, Smith JW (2002). Alcohol seeking by rats: action or habit? Q J Exp Psychol B 55: 331–348.
Drews E, Zimmer A (2010). Modulation of alcohol and nicotine responses through the endogenous opioid system. Prog Neurobiol 90: 1–15.
Gerdeman GL, Ronesi J, Lovinger DM (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5: 446–451.
Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr et al (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250: 1429–1432.
Iremonger KJ, Bains JS (2009). Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. J Neurosci 29: 7349–7358.
Koos T, Tepper JM, Wilson CJ (2004). Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci 24: 7916–7922.
Luk KC, Sadikot AF (2001). GABA promotes survival but not proliferation of parvalbumin-immunoreactive interneurons in rodent neostriatum: an in vivo study with stereology. Neuroscience 104: 93–103.
Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP et al (2012). Dichotomous organization of the external globus pallidus. Neuron 74: 1075–1086.
Mastro KJ, Bouchard RS, Holt HA, Gittis AH (2014). Transgenic mouse lines subdivide external segment of the globus pallidus (GPe) neurons and reveal distinct GPe output pathways. J Neurosci 34: 2087–2099.
Mathur BN, Capik NA, Alvarez VA, Lovinger DM (2011). Serotonin induces long-term depression at corticostriatal synapses. J Neurosci 31: 7402–7411.
Mathur BN, Tanahira C, Tamamaki N, Lovinger DM (2013). Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat Neurosci 16: 1275–1283.
Nielsen CK, Simms JA, Li R, Mill D, Yi H, Feduccia AA et al (2012). Delta-opioid receptor function in the dorsal striatum plays a role in high levels of ethanol consumption in rats. J Neurosci 32: 4540–4552.
Scherrer G, Tryoen-Tóth P, Filliol D, Matifas A, Laustriat D, Cao YQ et al (2006). Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Pro Natl Acad Sci USA 103: 9691–9696.
Schlösser B, Klausa G, Prime G, Ten Bruggencate G (1999). Postnatal development of calretinin- and parvalbumin-positive interneurons in the rat neostriatum: an immunohistochemical study. J Comp Neurol 405: 185–198.
Surmeier DJ, Song WJ, Yan Z (1996). Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci 16: 6579–6591.
Tepper JM, Koós T, Wilson CJ (2004). GABAergic microcircuits in the neostriatum. Trends Neurosci 27: 662–669.
Volpicelli JR, Alterman AI, Hayashida M, O'Brien CP. (1992). Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry 49: 876–880.
Wamsteeker Cusulin JI, Füzesi T, Inoue W, Bains JS (2013). Glucocorticoid feedback uncovers retrograde opioid signaling at hypothalamic synapses. Nat Neurosci 16: 596–604.
Wassum KM, Cely IC, Maidment NT, Balleine BW (2009). Disruption of endogenous opioid activity during instrumental learning enhances habit acquisition. Neuroscience 163: 770–780.
Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE et al (2014). Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology 39: 579–594.
Yin HH, Knowlton BJ, Balleine BW (2004). Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci 19: 181–189.
Yin HH, Ostlund SB, Knowlton BJ, Balleine BW (2005). The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci 22: 513–523.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Patton, M., Roberts, B., Lovinger, D. et al. Ethanol Disinhibits Dorsolateral Striatal Medium Spiny Neurons Through Activation of A Presynaptic Delta Opioid Receptor. Neuropsychopharmacol 41, 1831–1840 (2016). https://doi.org/10.1038/npp.2015.353
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.353
This article is cited by
-
The development of compulsive coping behavior depends on dorsolateral striatum dopamine-dependent mechanisms
Molecular Psychiatry (2023)
-
Compulsive alcohol consumption is regulated by dorsal striatum fast-spiking interneurons
Neuropsychopharmacology (2021)
-
TrkB-dependent disinhibition of the nucleus accumbens is enhanced by ethanol
Neuropsychopharmacology (2019)
-
Ensemble encoding of action speed by striatal fast-spiking interneurons
Brain Structure and Function (2019)
-
Acute drug effects on habitual and non-habitual responding in crossed high alcohol preferring mice
Psychopharmacology (2018)
