
Abstract
Posttraumatic stress disorder (PTSD) is both a prevalent and debilitating trauma-related disorder associated with dysregulated fear learning at the core of many of its signs and symptoms. Improvements in the currently available psychological and pharmacological treatments are needed in order to improve PTSD treatment outcomes and to prevent symptom relapse. In the present study, we used a putative animal model of PTSD that included presentation of immobilization stress (IMO) followed by fear conditioning (FC) a week later. We then investigated the acute effects of GR receptor activation on the extinction (EXT) of conditioned freezing, using dexamethasone administered systemically which is known to result in suppression of the HPA axis. In our previous work, IMO followed by tone-shock-mediated FC was associated with impaired fear EXT. In this study, we administered dexamethasone 4 h before EXT training and then examined EXT retention (RET) 24 h later to determine whether dexamethasone suppression rescued EXT deficits. Dexamethasone treatment produced dose-dependent enhancement of both EXT and RET. Dexamethasone was also associated with reduced amygdala Fkbp5 mRNA expression following EXT and after RET. Moreover, DNA methylation of the Fkbp5 gene occurred in a dose-dependent and time course-dependent manner within the amygdala. Additionally, we found dynamic changes in epigenetic regulation, including Dnmt and Tet gene pathways, as a function of both fear EXT and dexamethasone suppression of the HPA axis. Together, these data suggest that dexamethasone may serve to enhance EXT by altering Fkbp5-mediated glucocorticoid sensitivity via epigenetic regulation of Fkbp5 expression.
Similar content being viewed by others


Introduction
In human populations, exposure to traumatic stress can precipitate fear-related disorders (eg, posttraumatic stress disorder, PTSD) (Kessler et al, 1995; American Psychiatric Association, 2013). A growing body of translational research has identified the interaction of several neurobiological mechanisms underlying the mammalian response to traumatic stress (for recent review, see (VanElzakker et al, 2014). For example, in rodents, activation of the hypothalamic-pituitary-adrenal (HPA) axis invokes stress-induced plasticity in limbic structures such as the amygdala (Roozendaal et al, 2009). In addition, genetic and epigenetic regulation within the HPA axis has been associated with the human condition of PTSD (Yehuda et al, 2010).
The FKBP5 gene encodes for the protein FK506 binding protein 5 (FKBP5), which regulates the sensitivity of the glucocorticoid receptor (GR) (Binder 2009), and is critically involved in HPA axis activity during responses to stress. The GR is a critical component of the mammalian stress response system and represents a primary binding site for cortisol (corticosterone in rodents). The FKBP5 protein interacts with heat shock protein 90 to reduce GR affinity, modulating cortisol activity and preventing translocation of the GR complex to the nucleus at basal levels and following stress (Pratt and Toft 1997; Denny et al, 2000) by decreasing GR sensitivity to cortisol and preventing its translocation to the nucleus. Interestingly, epigenetic modifications of FKBP5 (and subsequently GR activity) have been found in psychiatric illness, including PTSD (Binder, 2009; Maddox et al, 2013). Furthermore, FKBP5 single nucleotide polymorphisms (SNPs) have been associated with more rapid feedback and enhanced HPA axis suppression using the dexamethasone (Dex; GR receptor agonist) suppression test (Binder et al, 2008; Klengel et al, 2013). This observation is consistent with differential FKBP5 regulation as a potential intermediate phenotype for trauma- and stress-related disorders.
A number of studies have also found FKBP5 genotype to be differentially associated with risk for developing PTSD and other psychiatric disorders following exposure to trauma (Mehta et al, 2011; Zannas and Binder, 2014). Four SNPs of the FKBP5 gene (rs9296158, rs3800373, rs1360780, and rs9470080) were found to interact with severity of child abuse as predictors of adult PTSD symptoms in urban, previously traumatized civilians (Binder et al, 2008). Additionally, it was found that the rs1360780 SNP moderates the risk for PTSD after early trauma and DNA methylation within functional glucocorticoid response elements of the FKBP5 gene (Klengel et al, 2013). Using structural MRI from traumatized female civilians, individuals who were homozygous for the rs1360780 T (‘risk’) allele demonstrated significantly lower fractional anisotropy (a measure of white matter integrity) in the left posterior cingulum (a pathway linked to increased expression of conditioned fear during extinction (EXT)) compared with individuals without the T allele (Fani et al, 2014). Finally, carriers of the rs1360780 T (‘risk’) allele also exhibited an attention bias toward threat compared with individuals without the T allele, along with alterations in hippocampal activation and morphology (Fani et al, 2013). Taken together with previous reports, it appears that hippocampal sensitivity to stressors and subsequent cortisol fluctuations leads to decreased hippocampal functioning and possibly reduced hippocampal volume. All these findings, along with a number of other recent observations (Xie et al, 2010; Zimmermann et al, 2011; Appel et al, 2011; Buchmann et al, 2014), suggest a model in which the FKBP5 genotype is associated with enhanced intermediate neural and behavioral phenotypes that may underlie increased risk for PTSD and other stress-related psychiatric illnesses following trauma exposure.
A potential mechanism of action of HPA axis dysregulation is GR hypersensitivity (Yehuda, 2009), as evidenced by exaggerated negative feedback in PTSD (Yehuda et al, 2002). Dysregulation of glucocorticoids and the HPA axis may differentially activate GR in PTSD patients as compared with healthy controls in a manner that can be detected by psychophysiological indices including fear-potentiated startle (Jovanovic et al, 2010). The SNP rs1360780 A within FKBP5 may be actually contributing to alterations in negative feedback of the HPA axis (Klengel et al, 2013). In a previous study of traumatized civilians with and without PTSD, we demonstrated that dexamethasone administration leads to a transient suppression of HPA function that may, in turn, normalize exaggerated fear in patients with PTSD (Jovanovic et al, 2010; Jovanovic et al, 2011). In the current study, we sought to determine whether controlled dexamethasone suppression would similarly enhance EXT of fear in a mouse model of stress-induced EXT deficits.
In rodents, it has been shown that chronic elevations of plasma corticosterone levels were associated with increased Fkbp5 expression and decreased fkbp5 methylation in blood and brain. Significant linear relationships were observed between DNA methylation of Fkbp5 and mean plasma corticosterone levels for both blood and brain following 4-week glucocorticoid exposure. The degree of methylation change in blood correlated significantly with both methylation and expression changes in hippocampus, with the notable observation that methylation changes occurred at different intronic regions when comparing blood vs brain tissue (Ewald et al, 2014). Furthermore, Fkbp5 methylation levels have been correlated with anxiety-like behavior (Lee et al, 2011). Also, brain regions with low basal Fkbp5 expression such as the amygdala display higher increases in Fkbp5 mRNA expression following stress as compared with regions with high basal expression, supporting the hypothesis that GR sensitivity is mediated via Fkbp5 (Scharf et al, 2011).
Using immobilization (IMO) stress in mice, we have previously demonstrated a marked increase in HPA axis activity after exposure to this stressor (Andero et al, 2011). In addition to an IMO-induced elevation in HPA axis activity, we observed impaired fear EXT and EXT retention (RET) following Pavlovian fear conditioning (FC) as a model of the fear-related symptoms of PTSD (Andero et al, 2011). Furthermore, IMO impaired long-term declarative memory and enhanced anxiety-like behavior (Andero et al, 2013). These approaches have contributed to translational evidence of previous trauma as a risk factor for PTSD via HPA-axis-related mechanisms (Mehta and Binder, 2012).
The current study was based on our hypothesis that Fkbp5 expression and GR regulation are critical factors in both FC and fear EXT memory consolidation. The goal of our study was to better understand the mechanisms by which corticosterone and Fkbp5 may mediate fear memory processing in the hippocampus and amygdala of animals previously exposed to a stressor shown to alter fear EXT and HPA axis regulation in mice (Andero et al, 2011).
Materials and Methods
Animals
All experiments were performed on adult (2–3 months old) wild-type C57BL/6J mice obtained from Jackson Labs. Male mice were group-housed in a temperature-controlled vivarium, with ad libitum access to food and water. They were maintained on a 12-h light/dark cycle, with all behavioral procedures being performed during the light cycle. All procedures used were approved by the Institutional Animal Care and Use Committee of Emory University and in compliance with National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Drugs
We administered dexamethasone (Sigma, St Louis, MO, USA, catalog no. D8893) intraperitoneally at a dose of 20 or 300 μg/kg in a vehicle of 15% dimethylsulfoxide in phosphate-buffered saline; the same vehicle was also used in control groups. In experiments in which dexamethasone was given, mice received a single dose 4 h before the appropriate behavioral procedure when levels of plasma corticosterone are believed to be at their greatest degree of suppression due to the drug (Jiang et al, 2009).
Immobilization Stress
IMO procedures were conducted in a room separate from housing and behavioral paradigms. Each animal was immobilized by gently restraining their four limbs in a prone position to metal arms attached to a wooden board for 2 h (Andero et al, 2011). All cage-mate animals received the same treatment: IMO or handling. Handling lasted ~1 min per mouse and consisted of letting the animal walk on top of their home cage and in the hands of the experimenter wearing latex gloves. After treatment, animals where returned to their home cage where they remained undisturbed until fear-learning procedures. All mice were habituated to the test chambers both before and after the IMO stress, receiving exposure to handling and the training context completely separate and distinct from the IMO context. The four handling sessions were performed 2 days before the IMO and 2 days before FC.
Cued FC and Fear EXT
Methods for Figures 2, 3 and 4
Mice were fear-conditioned and tested for levels of conditioned fear expression/ EXT in standard rodent modular test chambers (ENV-008-VP; Med Associates Inc, St Albans, VT) with an inside area of 30.5 cm (L) × 24.1 cm (W) × 21.0 cm (H). The tone-conditioned stimulus (CS) was generated by a Tektronix function generator audio oscillator delivered through a high-frequency speaker (Motorola, Model 948) attached to the side of each chamber. The illumination was provided by white light. Mice received five trials of a CS (30 s tone, 6 kHz, 70 dB) co-terminating with a footshock (500 ms, 1 mA) unconditioned stimulus (US). The PreCS and intertrial interval (ITI) were 180 s. The apparatus was cleaned with Quatricide after each mouse. Fear EXT and fear EXT RET was assessed 24 and 48 h, respectively, after FC and consisted of a PreCS period of 300 s, 50 CS tone trials (30 s each) with a 60 s ITI in the same chamber but changing the context and odor. The context was changed by covering the grids with Plexiglass and illuminating the chamber only with red light. The odor was altered by cleaning the apparatus with 70% EtOH after each animal. Tone presentation and freezing data were controlled, stored, and analyzed with FreezeView software (Coulbourn Instruments, Whitehall, PA) (Andero et al, 2011).
Startle Chamber and Fear EXT
Methods for Figure 1

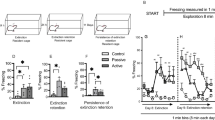
Previous exposure to immobilization (IMO) on wooden board causes impaired fear extinction (EXT) and enhanced corticosterone levels after both fear conditioning (FC) and EXT. (a) Half of the mice had exposure to IMO while the other half (Controls) had similar levels of handling but no IMO. Six days later, half of the mice of each group had cued FC. Only mice that received cued FC showed increased levels of freezing, that was similar in control vs IMO groups (***p⩽0.001) (n=46–48 per group). (b) Corticosterone levels were measured immediately after FC (0), as well as 30 and 120 min after finishing conditioning (R30 and R120, respectively) in different cohorts of mice. Mice that had a previous IMO showed enhanced levels of corticosterone after mere exposure to the shock chamber (time 0) vs non-IMO animals, revealing sensitization; after FC, enhanced corticosterone response was found at 0 and R30 in controls, and impaired recovery was found in previously immobilized mice at R30. +p<0.05 vs corresponding 0 time values; **p<0.01 vs corresponding 0 and R30 values; +++p<0.001 vs corresponding 0 time values; ***p<0.001 vs corresponding 0 and R30 values; a and b indicate comparisons are made between groups at the same sampling time (n=8 per group). (c) Twenty-four hours after FC, half of the mice had exposure to EXT, whereas the other half had exposure only to the experimental box. All mice that were exposed to conditioned cues (EXT) presented higher levels of freezing (IMO-Ext and Control-Ext vs IMO-NoEXT and Control-NoEXT). Additionally, the mice that had a previous exposure to IMO showed impaired EXT when compared with control mice. **p⩽0.01 vs Control-EXT, ***p⩽0.001 vs all other groups (n=24 per group). (d) Mice that had a previous IMO presented overall enhanced levels of corticosterone after EXT. Moreover, they also presented similar levels of corticosterone levels at 0 and R30, whereas at this time point, all other groups presented significantly lower levels at R30 compared with 0. +p<0.05 vs 0, ++p<0.01 vs 0, +++p<0.001 vs 0, **p<0.01 vs 0 and 30 (n=8 per group).
FC conditioning was conducted in specialized startle-footshock chambers (SRLAB, San Diego Instruments, San Diego, CA) consisting of a nonrestrictive acrylic plastic cylinder, (5.5 cm in diameter × 13 cm long) mounted on a Plexiglas platform that was located in a ventilated, sound-attenuated chamber. The footshock unconditioned stimulus (US) was delivered through a removable stainless steel grid floor using one of the four constant current shock generators (SDI, San Diego, CA) located outside the isolation chambers. A piezoelectric accelerometer mounted under each platform detected cylinder movements that were digitized and stored by an interfacing computer assembly. The tone CS was generated by a Tektronix function generator audio oscillator (Model CFG253, Beaverton, OR) and delivered through a high-frequency speaker (Motorola, Model 948). Sound intensities were measured with an audiometer (Radio Shack, Ft. Worth, TX, #33-2055). Stimuli presentation and data acquisition were controlled, digitized, and stored by an interfacing IBM PC compatible computer using SRLAB software. On each of the 2 days prior to training, mice were given a 10-min startle chamber exposure session to habituate mice to handling and the training context. During cued FC, mice received five trials of a CS tone (30 s, 6 kHz, 70 db) co-terminating with a footshock US (500 ms, 0.6 mA). The PreCS period and the ITI were 5 min. The experimental procedure was similar as previously described (Andero et al, 2011). The fear EXT test was performed as previously described in the paragraph above in the Med Associates chambers but using 30 CS.
Radioimmunoassays
Mice were killed at appropriate time points and trunk blood was collected in tubes containing ethylenediaminetetraacetic acid. All samples to be statistically compared were run in the same assay to avoid inter-assay variability. Plasma corticosterone levels (Figure 1) were determined by double-antibody radioimmunoassay as previously described (Andero et al, 2012). Corticosterone radioimmunoassay used 125I-corticosteronecarboximethyloximetyrosine-methyl ester (ICN-Biolink 2000, Barcelona, Spain), synthetic corticosterone (Sigma, Barcelona, Spain) as the standard, and an antibody raised in rabbits against corticosteronecarboximethyloxime-BSA kindly provided by Dr G. Makara (Institute of Experimental Medicine, Budapest, Hungary). Plasma corticosteroid-binding globulin was inactivated by low pH). Plasma corticosterone levels illustrated in Figure 2d were determined with the MP BIOMEDICALS Corticosterone Double Antibody RIA Kit #07120102. Procedure was followed as indicated by the manufacturer. This kit has no cross-reaction with dexamethasone.

Dexamethasone enhances the extinction of learned fear. (Top) Timeline of the experimental procedures. All mice were exposed to immobilization to a wooden board (IMO) for 2 h, 6 days prior to fear conditioning. Mice were trained for cued fear conditioning with five tone-shocks pairings (conditioned stimulus; CS). Four hours before EXT mice received vehicle or dexamethasone treatment: (Veh) (n=42), dexamethasone 20 μg/kg (Dx 20) (n=43), or dexamethasone 300 μg/kg (Dx 300) (n=23), all given systemically (i.p.). (a) Freezing during fear conditioning. All groups presented similar levels of freezing during habituation (pre-conditioned stimulus; Pre-CS) and during fear conditioning. (b) Four hours before fear extinction learning (EXT), vehicle or dexamethasone (20 and 300 μg/kg i.p.) was given, and mice were exposed to trials of CS without US presentations. The dexamethasone 300 μg/kg group presented significantly less freezing than the vehicle group during conditioned stimulus presentation; CS1–50 (*p<0.05). (c) Freezing levels are shown during extinction retention (RET) 24 h later, with no drug on board (n=7–10 per group). Mice that received dexamethasone 20 μg/kg during EXT now had significant lower levels of freezing in RET than the animals that had vehicle during CS1–50 (**p<0.01). (d) Blood corticosterone levels are shown following home cage, or EXT or RET exposure. Corticosterone levels were higher in the Veh group after EXT than HC group (*p<0.05). Two hours after RET, the mice receiving dexamethasone 20 μg/kg show less corticosterone than two hours after EXT (**p<0.01). Two hours after RET, dexamethasone 300 μg/kg show higher levels of corticosterone than two hours after EXT (#p=0.055). (e) During EXT, the dexamethasone treatment at different doses (20 or 300 μg/kg) did not alter freezing during the intertrial interval (ITI). (f) During RET, the dexamethasone 20 μg/kg group shows more discrimination (less freezing) than vehicle group in the ITI of the period between stimulus (*p<0.05). (g) The mice that received dexamethasone 20 μg/kg had significant lower average levels of freezing overall than the animals that had vehicle during RET (*p<0.05). (h) Fkbp5 messenger RNA (mRNA) gene expression was determined in the amygdala at 2 h after EXT. The 20 μg/kg group (n=17) had significantly decreased levels of Fkbp5 mRNA when compared with Vehicle (n=14) and 300 μg/kg (n=9) groups (***p<0.001). The 300 μg/kg group also presented enhanced Fkbp5 mRNA levels than Vehicle (***p<0.001). (i) Fkbp5 messenger mRNA gene expression was determined in the amygdala at 2 h after RET. The 20 μg/kg (n=8) group had significantly decreased levels of Fkbp5 mRNA when compared to Vehicle (n=10) (*p<0.05) and the 300 μg/kg group (n=7). (j) The mice that received dexamethasone 20 μg/kg had significant lower average level of freezing than the animals that had vehicle during ITI in RET (*p<0.05). (k) Fkbp5 mRNA levels in the hippocampus after EXT. The group 300 μg/kg (n=6) presented enhanced levels of Fkbp5 mRNA compared with vehicle (n=6) and 20 μg/kg (n=6) (***p<0.001). (l) There were no statistical differences in Fkbp5 mRNA levels in the hippocampus after RET.
Reverse Transcription and PCR Quantification
Total RNA was isolated with Qiagen RNeasy kit (Qiagen, Valencia, California) (Lee et al, 2011). Gene expression changes in the amygdalar and hippocampal tissue were detected by relative quantitative reverse transcription PCR (FAST 7500; Applied Biosystems, Foster City, CA). Bilateral tissue punches were performed based on the Mouse Brain Atlas by Paxinos (Paxinos and Franklin, 2006). At the time the mice were killed, brains were placed on ice and rapidly blocked, and tissue punches were obtained for isolation of messenger RNA (mRNA) and DNA. Total RNA was reverse-transcribed using the RT2 First Strand Kit catalog # 330401 (Qiagen) according to the manufacturer’s instructions. TaqMan assays (Applied Biosystems, Carlsbad, CA) were used to quantify the expression of Fkbp5 (Mm00487401), Dnmt1 (Mm01151063), Dnmt3a (Mm00432881), Dnmt3b (Mm01240113), Tet1 (Mm011690870), Tet2 (Mm00524395), and Tet3 (Mm00805756) normalized to mouse Gapdh (4352932E). Relative levels of mRNA expression were normalized in all the samples with expression levels of glyceraldehyde- 3-phosphate dehydrogenase (GAPDH) (Maguschak and Ressler, 2011). Graphics are represented by fold change obtained with the 2^-ddCt method.
DNA Methylation Analyses
DNA methylation at functional glucocorticoid response elements in mouse Fkbp5 intron 5 described by Lee et al (2010) was performed based on procedures described by Sabbagh et al (2014), using Varionostic GmbH, Inc (Ulm, Germany, www.varionostic.de). Briefly, 500 ng genomic DNA was bisulfite-converted using the EZ-96 DNA methylation kit (Zymo, Irvine, CA). A 412-bp fragment was amplified from bisulfite-converted DNA using the primers F_mouse-I5 5′-aggaagagagAATATTTTGTTTTGAATGTGGTTGG-3′ and R_mouse-I5 5′-cagtaatacgactcactatagggagaaggctCTCTCCTAAAAACCCTTACCCAATA-3′ followed by Sequenom EpiTYPER DNA methylation analysis performed according to the instructions of the manufacturer. Uncertainty threshold was set to 0.2.
Statistical Analysis
Statistics were performed with IBM SPSS Statistics Version 19.0 (Armonk, NY) and GraphPad Prism 6.05 for Windows (GraphPad Software, La Jolla, CA). Detection of outliers was performed and removed from analyses when necessary. The criterion was more than 2 SD below or above the average. Repeated-measures ANOVA, one- or two-way ANOVA, or Student’s t test (two-tailed) for independent samples were used where appropriate. In all cases, if a statistically significant interaction was found, additional comparisons were calculated. Bonferroni post hoc analyses were performed where appropriate. The corticosterone levels were analyzed with Generalized Linear Models, Generalized Estimating Equations models. The significance of the effects was determined by the Wald χ2 statistic. In order to investigate the association between Fkbp5 expression, freezing in EXT, Dnmt and Tet mRNA expression, and Fkbp5 intron 5 methylation, we ran bivariate Pearson correlations between Fkbp5 expression (fold change), freezing in EXT (spent time %), Dnmt and Tet mRNA expression (fold change), intron 5 methylation (%), and Tet expression (Fold change). The alpha level was 0.05. We also examined these correlations separately in the Vehicle group and dexamethasone-treated groups. The results are presented as means±or+SEM, and statistical significance was set at P<0.05.
General Procedures for Figures 2, 3 and 4

Correlations between Fkbp5, Dnmt, and Tet mRNA expression in the amygdala and freezing in fear extinction learning (EXT) and fear extinction retention (RET). (a) In EXT between Fkbp5 and freezing, a significant positive correlation was found in vehicle group (Veh) (R=0.60, p<0.05, n=14), but not in the dexamethasone 20 μg/kg group (Dx 20) (R=−0.29, n=17); moreover, a significant negative correlation was found in the dexamethasone 300 μg/kg group (Dx300) (R=−0.73, p<0.05, n=9). (b) In RET, between Fkbp5 and freezing, a strong positive correlation was found in the dexamethasone 300 μg/kg group (R=0.81, p<0.05, n=7). There were no significant correlations in the vehicle (R=−0.07, n=10) nor 20 μg/kg group (R=0.59, n=8) (c) A significant positive correlation was found between Fkbp5 and Dnmt3b in the amygdala at 2 h after EXT (R=0.70, p<0.0001, n=29). (d) A positive correlation was also found between Fkbp5 and Tet2 in the amygdala at 2 h after EXT (R=0.46, p<0.01, n=30).

Dexamethasone induced changes in Fkbp5 DNAm and Dnmt and Tet mRNA expression in the amygdala after fear extinction learning. (a) Dexamethasone induced dose-dependent time-course alteration of methylation on intron 5 CpG4 of Fkbp5 in the amygdala. Two hours after fear extinction learning (EXT), dexamethasone 20 μg/kg group (Dx20) showed higher DNAm than the 300 μg/kg group (Dx300) (*p<0.05, n=7–11 per group). But this is not found in CpG 5 (b, n=7–13 per group). Dexamethasone-induced changes are dose-dependent in a time-course downregulation and up-regulation of Dnmt (c–e), Tet (f–h) (n=7–13 per group). Dnmt1 was not found to change (c), but dexamethasone 20 μg/kg was associated with less Dnmt3a (d) (*p<0.05 vs vehicle group (Veh)) and Dnmt3b expression (e) (*p<0.05 Dx20 vs Dx300). The dexamethasone 300 μg/kg group presented higher levels of Tet2 expression 2 h after EXT (g) (*p<0.05 vs Veh and Dx20). Moreover, there are significant differences between 2 and 6 hrs after EXT in Tet1 (f) and Tet3 (h) (**p<0.01, ***p<0.001).
Mice were exposed to IMO or received compensatory handling. Six days later, the mice were exposed to FC and corticosterone levels were measured immediately after, 30 and 120 min after FC. Twenty-four hours later, a different cohort of mice received a dose of dexamethasone (vehicle, 20 and 300 μg/kg) 4 h before EXT. One group of mice were killed to determine corticosterone levels 30 min after EXT, whereas another group was used to measure amygdala and hippocampus Fkbp5 mRNA levels and amygdala Fkbp5 methylation levels 2 h after EXT. Another cohort of mice received a session of RET 24 h after EXT and were killed 2 h after RET to determine amygdala and hippocampus Fkbp5 mRNA levels.
Results
IMO Stress Effects on FC, EXT, and Corticosterone Levels
Six days following IMO procedures, animals were either subjected to FC (FC groups) or were placed in the conditioning box without receiving CS or US presentations (NoFC groups) (Figure 1a). All groups presented similar levels of freezing before the CS was presented during the habituation to the FC apparatus. FC robustly lead to freezing behavior during the CS presentation compared with control treatment (repeated measures (rm) ANOVA F1,184=1263.751 p<0.0001) without differences between IMO and non-IMO groups.
In a parallel group of animals, we examined corticosterone levels immediately, 30 and 120 min after FC (Figure 1b). The sampling Time factor, Wald χ2 (2)=255.469 p<0.001, and Group (FC or NoFC) factor, Wald χ2 (1)=9.911 p<0.01, were significant, suggesting that corticosterone levels changed with the passage of time as well as in response to FC. Moreover, there were interactions between Group and Time which demonstrates that fear-conditioned mice exhibited a different time course of corticosterone levels as compared with NoFC animals, Wald χ2 (2)=28.287 p<0.001, and IMO × Group × Time, Wald χ2 (2)=7.220 p<0.05. Pairwise comparisons showed that all groups at the 120-min time displayed significant reduction in corticosterone when compared with the 30-min and 0-min (immediate) time points (p<0.001 IMO—NoFC, Control—FC, IMO—FC, and p<0.01 Control—NoFC). At 30 min, the group IMO—NoFC and Control—FC groups showed significant differences vs 0 min, p<0.001 and p<0.05, respectively. However, neither Control-NoFC nor IMO-FC group showed significant reductions in corticosterone at 30 min after conditioning. The 30 min time point data suggests that FC enhanced corticosterone levels in IMO mice compared with mice that were not exposed to IMO. Then, pairwise comparisons between groups were made at the same time point showing that the FC groups, independently of IMO/Control condition, showed higher levels of corticosterone at 30 min, p<0.05. Moreover, the Control—NoFC group had lower levels at time 0 than all other groups, p<0.05. At 120 min, all groups displayed similar levels of corticosterone, which suggests a return to baseline levels.
Additional groups of mice underwent EXT, and freezing levels were measured (Figure 1c). All groups presented similar levels of freezing during the habituation to the conditioning apparatus during the EXT session. Animals previously exposed to IMO exhibited enhanced levels of freezing during EXT (IMO factor, rmANOVA F1,92=13.158 p<0.0001; IMO × FC interaction, rmANOVA, F1,188=13.091 p<0.0001). Simple effects tests showed that the IMO—EXT and Control—EXT groups froze significantly longer than the IMO—NoEXT and Control—NoEXT groups (p⩽0.0001 in CS1–10, CS11–20 and CS21–30). Moreover, the IMO—EXT group showed increased levels of freezing during EXT when compared with the Control—EXT group (p<0.001 CS1–10, CS11–20 and p<0.0001, CS21–30).
Animals were killed following EXT to assess plasma corticosterone levels (Figure 1d). The Time factor was significant, Wald χ2 (2)=91.017 p<0.001, and there was an interaction between IMO and Group (EXT/NoEXT) Wald χ2 (1)=5.417 p<0.05 and IMO and Time Wald χ2 (2)=6.558 p<0.05. This suggests that plasma corticosterone levels changed at different time points, in general, and that IMO differently affected plasma corticosterone levels in EXT and noEXT. Moreover, IMO changed the plasma corticosterone levels evaluated at different time points. Pairwise comparisons showed that IMO—NoEXT Groups vs IMO—EXT, p=0.077, showed marginally significant differences in mice that were fear-extinguished vs those that were not extinguished. Plasma corticosterone was significantly different in the Control—NoEXT and IMO—NoEXT groups, at both 30 and 120 min, as compared with the immediate (0 min) time point, (120 vs 0 min, p<0.001) except IMO—NoEXT (30 vs 0 min, p<0.05). Interestingly, plasma levels in the Control—EXT group were lower as compared with the IMO—EXT group (p=0.011) showing that a history of previous IMO stress enhances corticosterone levels after EXT. This is consistent with significant differences observed between 120 vs 30 min and 0 min (immediate) groups (p<0.01, in the IMO—EXT group, whereas there are significant differences at both 120 and 30 min vs 0 (30, p<0.01 and 120, p<0.001). This suggests that the IMO mice have a slower HPA axis recovery after EXT than mice that had no previous IMO exposure. An alternative interpretation may be that that the enhanced corticosterone levels are the result of impaired EXT with consequently higher levels of freezing and other fear-related measures, including the HPA axis.
Dexamethasone Administration Produces a Dose-Dependent Enhancement of Fear EXT Learning and EXT RET in IMO Mice
Dexamethasone pretreatment has been shown to modulate fear learning in PTSD patients (Jovanovic et al, 2010; Jovanovic et al, 2011). Thus, we hypothesized that it may enhance fear EXT learning and RET in our model of PTSD fear-related symptoms. For this reason, we examined the effects of low vs high dexamethasone administration (20 vs 300 μg/kg) compared with vehicle on EXT and RET in mice exposed to IMO 6 days before FC. Mice in all groups showed no differences in the levels of FC prior to dexamethasone injection (Figure 2a).
We then administered vehicle or dexamethasone (20 or 300 μg/kg) to IMO mice 4 h before EXT in the morning, and collected behavioral data during EXT (Figure 2b and e). All groups presented similar levels of freezing during the habituation to the experimental box. Analysis of freezing during EXT revealed that dexamethasone-treated (300 μg/kg) animals had significantly lower levels of freezing than vehicle-treated animals (rmANOVA, F2,105=3.511 p<0.05). The high-dose dexamethasone (300 μg/kg) group froze significantly less than the vehicle group (multiple comparisons, p<0.05) during within-session EXT (Figure 2, panel b). There were no statistical differences in the ITI freezing levels during EXT.
Twenty-four hours after EXT in the absence of any drug, we examined freezing levels during EXT RET (Figure 2c, f, g, and j). Animals that received low-dose dexamethasone (20 μg/kg) displayed significantly lower levels of freezing than vehicle-treated animals (rmANOVA F2,22=6.401 p<0.01; Figure 2c). In addition, the dexamethasone (20 μg/kg) group froze significantly less than the vehicle group in CS1–50 (multiple comparisons, p<0.05; Figure 2c). During RET, the dexamethasone (20 μg/kg) group also froze significantly less than the vehicle group between CS presentations across the RET session (rmANOVA F2,22=6.519 p<0.01, multiple comparisons p<0.05; Figure 2f). Furthermore, the dexamethasone (20 μg/kg) group showed less total average freezing during the CS (p<0.05, one-way ANOVA, F2,22=6.395, followed by Bonferroni post hoc test p<0.05; Figure 2g) and during the ITI, a measure of fear generalization (p<0.01, one-way ANOVA, F2,22=6.577, followed by Bonferroni post hoc test p<0.05; Figure 2j).
In summary, although the high dexamethasone dose may enhance apparent within-session EXT, this effect is not maintained during EXT RET testing off of the drug. In contrast, the low dexamethasone dose enhances the consolidation of fear EXT.
Dexamethasone Dose-Dependently Modulates Corticosterone Levels after Fear EXT and Fear EXT RET
Dexamethasone dose-dependently modulated the corticosterone levels after EXT and RET when administered 4 h before EXT in mice with a previous IMO (Figure 2d). Corticosterone levels were higher in the vehicle-treated FC group after EXT training (EXT) than the handled control in home cage group (t=3.436 df=6, p<0.05), but there was not a significant reduction in corticosterone levels following EXT RET. In contrast, in the low-dose dexamethasone group (20 μg/kg), there was a significant reduction in plasma corticosterone levels after RET which suggests reduced HPA axis activity associated with their decreased fear expression during RET (t=2.949, df=16, p<0.01). In the highest dose dexamethasone group (300 μg/kg), an opposite effect was seen, with decreased corticosterone following EXT training but increased corticosterone levels after RET (t=2.092, df=14, p=0.0551, Figure 2d). Interestingly, although the high dose of dexamethasone was associated with more corticosterone suppression during EXT, there was an apparent u-shaped curve, such that the high dose was not associated with greater EXT RET.
Dexamethasone Modulates Fkbp5 mRNA Expression in the Amygdala and Hippocampus of IMO Mice after Fear EXT and Fear EXT RET
Basal levels of Fkbp5 mRNA in the amygdala are similar to home cage group when evaluated 6 days after IMO (Supplementary Figure 2). Dexamethasone administration (20 and 300 μg/kg) 4 h before EXT induced dose-dependent alterations of Fkbp5 mRNA expression in the amygdala, associated with cued fear, after EXT (Figure 2h) and RET (Figure 2i) as well as in the hippocampus, associated with fear generalization (Figure 2k and l), respectively.
The lower-dose dexamethasone (20 μg/kg)-treated groups showed Fkbp5 mRNA downregulation in the amygdala after EXT (one-way ANOVA F2,37=20.70, p<0.001, followed by Bonferroni post hoc test, 20 μg/kg vs vehicle, p<0.001; Figure 2h). In contrast, the high-dose dexamethasone (300 μg/kg) group exhibited an upregulation of Fkbp5 mRNA expression in the amygdala (p<0.001 followed by Bonferroni post hoc test). Dexamethasone (300 μg/kg) administration also induced the upregulation of Fkbp5 mRNA expression in hippocampus after EXT (one-way ANOVA F2,15=21.76, p<0.001, followed by Bonferroni post hoc test Veh vs Dex300 p<0.001, Figure 2k).
We then examined Fkbp5 mRNA levels after RET in IMO mice. The Fkbp5 mRNA levels were again examined in the amygdala (Figure 2i) and hippocampus (Figure 2l). The dexamethasone (20 μg/kg) showed a downregulation of Fkbp5 mRNA in the amygdala after RET (one-way ANOVA F2,22=3.750, p<0.05, followed by Bonferroni post hoc test, Veh vs Dex20 p<0.05, Figure 2i). There were no significant effects of dexamethasone treatment and Fkbp5 levels in the hippocampus after RET (Figure 2l). In summary, a high dose of dexamethasone elicited an upregulation of Fkbp5 mRNA expression after EXT and a low dose of dexamethasone produced a downregulation of Fkbp5 mRNA expression after EXT and RET. Thus, the dose of dexamethasone associated with enhanced behavioral measures of EXT was also associated with downregulation of Fkbp5 in the amygdala during both EXT and RET.
Changes in Methylation of Fkbp5 in IMO Mice Following Dexamethasone Administration before Fear EXT
DNA methylation can be dynamically regulated and may serve as a long-lasting mechanism to regulate gene expression in response to environmental stimulation. We therefore hypothesized that dexamethasone administration would alter Fkbp5 DNA methylation leading to an alteration of Fkbp5 expression. We investigated DNA methylation in and around a functional GR binding site in intron 5 of Fkbp5 previously shown to be responsive to corticosterone administration (Lee et al, 2010). Sequenom Epityper was performed at six CpG dinucleotides in intron 5 of the mouse Fkbp5 gene in the amygdala of mice treated with dexamethasone and compared with those of vehicle-treated controls at 2 and 6 h after EXT (Figure 3a and b). Fkbp5 DNA methylation is increased in intron 5 at CpG4 that is within a glucocorticoid response element (Supplementary Figure 1) 2 h after EXT in dexamethasone-treated mice when comparing the low dose (20 μg/kg) with the high dose (300 μg/kg) (two-way ANOVA F2,47=4.454, p<0.05, followed by Bonferroni post hoc test; p<0.05, Figure 3a). However, there was no significant difference at CpG5 (Figure 3b). These data suggest that dexamethasone enhancement of EXT is associated with increased DNA methylation at CpG4 intron 5 of Fkbp5, in a dose- and time-dependent manner within the amygdala. Increased Fkbp5 DNA methylation would be expected to associate with decreased Fkbp5 mRNA expression following EXT as found above.
Dexamethasone Modulates Methylation and Hydroxymethylation Markers after Fear EXT
We next examined whether enzymes involved in the dynamic alteration of DNA methylation are altered with fear EXT and dexamethasone treatment. Ten-eleven translocation (TET) proteins (Kohli and Zhang 2013) and the enzyme, DNA methyltransferase (DNMT) (Jaenisch and Bird 2003; Goll and Bestor 2005), have an important role in regulating DNA methylation. Specifically, TET proteins mediate the hydroxymethylation of DNA, whereas DNMT is responsible for DNA methylation (Gavin et al, 2013). It was previously observed that methylation alteration after corticosterone administration correlated with Dnmt regulation (Yang et al, 2012).
In the present study, dexamethasone induced dose- and time-dependent changes in Dnmt (Figure 3c–e) and Tet mRNA expression in the amygdala after EXT (Figure 3f–h). Dnmt1 expression was not changed; however, there was an effect of time and dose on Dnmt3a mRNA expression (F1,53=8.383 p<0.01) with low-dose dexamethasone (20 μg/kg) showing lower levels of expression compared with the vehicle group 2 h after EXT (p<0.05; Figure 3d). Dnmt3b mRNA expression was also significantly affected by dexamethasone (F2,53=3.328, p<0.05) following a similar pattern, with the low-dose group showing less expression than the high-dose group 2 h after EXT (p<0.05; Figure 3e). Tet1 mRNA expression was also time-dependent with reduced expression observed at 6 h, as compared with 2 h, after EXT (F1,53=11.82 p<0.01; Figure 3f). There was a significant effect of time (F1,53=10.95 p<0.01) and drug (F2,53=4.254 p<0.05; Figure 3g) in Tet2 expression. The high-dose (300 μg/kg) group showed more expression as compared with the vehicle and low-dose (20 μg/kg) groups 2 h after EXT (p<0.05). There was also a significant effect of time in Tet3 mRNA expression (F1,53=14.10 p<0.001, Figure 3h). Together, these data show that the epigenetic machinery needed for the alteration of DNA methylation is dynamically changing, in a treatment-dependent manner, during the EXT of conditioned fear, providing a potential mechanism for the transient increase in methylation and decrease in expression of Fkbp5.
Correlations between freezing during the EXT and RET sessions and Fkbp5, Dnmt, and Tet mRNA expression in the amygdala were examined and are shown in Figure 4. In the vehicle group, there is a strong positive correlation between Fkbp5 mRNA expression in the amygdala and freezing during CS 21–30 of EXT training, representing the epoch of EXT where the most individual differences are found when analyzing correlations (R=0.60, p<0.05; Figure 4a), but not in the low-dose dexamethasone (20 μg/kg) group (R=−0.29, p>.05; Figure 4a). In addition, there is a strong negative correlation in the high-dose dexamethasone (300 μg/kg) group (R=−0.73, p<0.05). There was a significant positive correlation between Fkbp5 and freezing during CS 21–30 of RET in the high dexamethasone-treated group (300 μg/kg, R=0.81, p<0.05; Figure 4b). Two hours after EXT, a strong positive correlation was found between Fkbp5 and Dnmt3b (R=0.70, p<0.0001, Figure 4c) and a positive correlation was found between Fkbp5 and Tet2 (R=0.46, p<0.01, Figure 4d). Together, these data suggest that both EXT of fear and the regulation of epigenetic regulatory machinery are significantly associated with Fkbp5 regulation as a function of HPA suppression (Figure 5).

Simplified schematic of the results. (1) I.p. injection of dexamethasone transiently decreases corticosterone levels. (2) Fkbp5 protein acts as an inhibitory chaperone preventing GR translocation to the nucleus. With increased corticosterone binding, Fkbp5 is displaced by Fkbp4, allowing for translocation and exertion of its action as a transcription factor. (3) GR translocation to the nucleus acts on Fkbp5 via its glucocorticoid response elements (GREs) regulating processes such as methylation. Fkbp5 methylation alters Fkbp5 mRNA levels (4) that, in turn, affect the Fkbp5 protein levels (5). (6) The Fkbp5 protein levels negatively correlate with the GR sensitivity.
Discussion
To our knowledge, this is the first study demonstrating that dexamethasone suppression prior to EXT training leads to dynamic Fkbp5 regulation in the amygdala and enhanced EXT. This is in line with previous reports showing that dexamethasone can enhance fear EXT in rats as indexed by fear-potentiated startle measures and contextual fear (Yang et al, 2006, 2007; Ninomiya et al, 2010). Together these animal models corroborate our findings in humans showing dexamethasone suppression of the HPA axis prior to fear EXT, in an EXT-deficit model, enhances EXT of conditioned fear.
First, we showed that a 2-h IMO stress administered 6 days before FC did not influence the acquisition of conditioned fear but led to impairment in fear EXT learning. This was accompanied by increased corticosterone plasma levels after FC and EXT training in animals that were exposed to IMO vs handled controls. A potential explanation for these findings may be hormonal sensitization after a stressful experience when animals were previously exposed to IMO (Belda et al, 2008a, 2008b, 2012; Gagliano et al, 2008). Of note, our data suggest that hormonal sensitization was also found in response to exposure to the FC chamber in the absence of shocks. In the current study, we suggest that the assessment of fear EXT in animals previously stressed by IMO can be considered as a translational model of the fear-related symptoms of PTSD. This is due to the nature of IMO as one of the most severe rodent stressors as indicated by several markers of stress intensity (Marquez et al, 2002) and previous reports of IMO-induced alterations in fear learning and deficits in fear EXT (Andero et al, 2011, 2013). IMO has face validity as an animal model of the fearful aspects of human PTSD because exposure to severe stress is a necessary precursor for the development of PTSD (Teicher and Samson, 2013). In addition, IMO possesses construct validity in that both human PTSD patients and severely stressed animals show alterations in the regulation of the HPA axis (Yehuda, 2009). Further, EXT impairments have been reported in clinical populations with fear-related disorders (Wessa and Flor 2007; Norrholm et al, 2011; Milad et al, 2013). Concordantly, a single IMO exposure also leads to a deficit in EXT of conditioned fear and enhanced anxiety-like behavior evaluated with the elevated plus maze (Andero et al, 2013). Other stress models also have shown that a single exposure to a significant stressor is sufficient to impair EXT (Izquierdo et al, 2006).
We also demonstrated that dexamethasone, a systemic GR agonist, enhances EXT learning. In humans, dexamethasone suppression has been reported to change after traumatic stress (Klaassens et al, 2010) and in traumatized individuals who develop PTSD symptoms (Yehuda et al, 2014). It has been reported that dexamethasone dose-dependently enhances EXT in fear-potentiated startle and contextual fear (Yang et al, 2006, 2007; Ninomiya et al, 2010). Our current data show that dexamethasone enhances EXT and that this enhancement is coupled to dynamic Fkbp5 regulation. The effect of dexamethasone on fear EXT in previously immobilized animals was dose-dependent and non-linear. Interestingly, while a high-dose dexamethasone (300 ug/kg) appeared to decrease fear expression while on board, in the EXT training session, it had no effect on EXT RET. In contrast, the low dose of dexamethasone (20 μg/kg) had less of an effect during EXT testing, but was most effective in enhancing EXT RET. The IMO-induced disruption of EXT is thought to be due to abnormalities in HPA axis function with alterations in FKBP5 dynamics (Klengel et al, 2013) including FKBP5 feedback. Our previous work has shown that FKBP5 ‘risk’ polymorphisms that manifest with enhanced induction of FKBP5, altered cortisol release, and impaired negative feedback are associated with an increased risk to develop PTSD or related symptoms. Consistent with the ultra-short negative feedback between the FKBP5 protein and GR activation, genotype-mediated increases in FKBP5 transcription have been shown to be associated with GR resistance (Moriceau et al, 2006; Binder et al, 2008) and with a decreased efficiency of negative feedback of the stress hormone axes accompanied by prolonged cortisol release (Ising et al, 2008). The SNPs, rs3800373 and rs1360780, have been associated with increased peri-traumatic dissociation in children after medical trauma (Koenen et al, 2005) and higher levels of peri-traumatic dissociation have been shown to be predictors of PTSD in adults (Ozer et al, 2003).
In our study, correlations between freezing in EXT and Fkbp5 dynamics revealed significant differences between mice treated with dexamethasone as compared with vehicle. After the EXT and RET sessions, the vehicle group showed both a high level of corticosterone and Fkbp5 expression whereas the low-dose dexamethasone-treated group displayed both low levels of corticosterone and Fkbp5 expression on RET; effects that may be related to the observed dexamethasone-induced enhancement of EXT learning. A large number of findings from the 1990s and later suggest that a peripheral low dose administration of dexamethasone is generally prevented from passing the blood–brain barrier. A major player in this process is the P-glycoprotein expressed by the multidrug resistance gene mdr1a (Smit et al, 1998). Given the opposite findings of the low- and high-dose dexamethasone treatment in the current study, an involvement of the blood–brain barrier or the P-glycoprotein seems likely. A potential explanation of our findings may be that the low dose of dexamethasone used in the study (20 μg/kg) is sufficient to fully occupy GRs at the periphery, including the pituitary, thereby effectively shutting down the HPA axis (re)activity and depleting the brain from endogenous GR agonists (native or synthetic) (Cole et al, 2000). This consequently results in decreased expression of Fkbp5. In contrast, with the high dose of dexamethasone (300 μg/Kg), which potentially penetrates the blood–brain barrier (Miller et al, 1992), the brain may be flooded with this agonist, thereby mimicking a strong stress response. This is also in line with the induction of Fkbp5 expression in several brain regions. Additionally, increased levels of Fkbp5 could impair brain GR action, particularly under conditions of blockade of endogenous corticosterone release, as it is actually observed during the EXT session of the high-dose group. Thus, two different doses of dexamethasone may result in very opposite effects, especially for the brain. Of note, when animals are tested for RET, most remaining dexamethasone has been eliminated from the system.
Evidence suggests that the role of FKBP5 in the mammalian stress response includes epigenetic mechanisms such as DNA methylation status. Previous reports from the clinical literature have shown that methylation of the Fkbp5 gene (at the promoter region of exon 1) did not predict treatment response, but decreased in association with recovery (Yehuda et al, 2013). A study using a chronic corticosterone administration paradigm, that had been previously shown to result in a phenotype similar to chronic stress exposure (Gourley et al, 2009), induced an increase in Fkbp5 mRNA expression in the hippocampus, hypothalamus, and blood of mice with a correspondent decrease in DNA methylation at Fkbp5 intron 5 (Lee et al, 2010).
Exposure to a FC paradigm (FC, EXT, RET) in animals previously stressed with IMO produces an acute elevation of plasma corticosterone levels (Andero et al, 2011). Corticosterone binds to GR with heat shock protein 90 (Hsp90) and the GR complex is translocated into the nucleus to initiate downstream genomic effects, a process thought to be inhibited by Fkbp5 binding. GR-mediated gene transcription leads to Fkbp5 upregulation accompanied by altered DNA methylation of Fkbp5. Increased Fkbp5 mRNA and protein expression are associated with impaired HPA feedback, and consequently, EXT of conditioned fear. Our data suggest that dexamethasone treatment rescues impaired EXT in a dose- and time-dependent manner. At a higher dose (300 μg/kg), dexamethasone initially appears to enhance within-session EXT when EXT learning occurs 4 h after dexamethasone injection. This suggests that the high dose of dexamethasone directly affects the amygdala to attenuate fear as an acute effect of HPA axis suppression. However, 28 h later, during RET, animals that received this high dose behaved no different from controls. Thus, this suggests that this very high-dose dexamethasone may drive additional GR activation instead of primarily GR suppression, increasing Fkbp5 expression levels in the hippocampus and amygdala.
In contrast, low dose of dexamethasone was associated with lower conditioned fear expression during the RET test which is indicative of a facilitatory effect on EXT. A potential explanation of the findings is that the low dose of dexamethasone may have caused a pharmacological suppression of adrenal activity for ~4 h and then resulting in decreased levels of Fkbp5 mRNA in the amygdala. Thus, the corticosterone released during EXT may have acted in a period of higher sensitivity to GR because of this downregulation of Fkbp5 levels. Our genomic findings suggest that low-dose dexamethasone induces downregulated Fkbp5 mRNA expression with alternations in Fkbp5 DNA methylation. The current findings highlight the dynamics of mRNA expression (upregulation/downregulation) associated with differential intronic methylation status, also associated with alterations in the Dnmt and Tet protein families. We obviously cannot discount the contribution of other neurobiological pathways on HPA axis activity and subsequent behavioral effects in fear EXT learning. Other candidate systems include, but are not limited to, brain-derived neurotrophic factor (Gourley et al, 2009), pituitary adenylate cyclase activating polypeptide (Stroth et al, 2011), the endogenous cannabinoid system (Hill and Tasker, 2012), and Nociceptin—Nociceptin receptor (Andero, 2015).
Our data suggest that low-dose dexamethasone administration can rescue impaired EXT learning produced by prior IMO stress in a model of the fear-related symptoms of PTSD. Further, these data also suggest that Fkbp5 upregulation may be associated with fear-related psychopathology and lend support to recent findings from a human study of FKBP5 polymorphisms and PTSD risk (Klengel et al, 2013). Clinical fear-related disorders appear to be related, in part, to EXT learning deficits and the subsequent ineffectiveness of EXT-based exposure therapies in treatment-resistant patients. These findings suggest further translational study of the dynamics of FKBP5 genomic regulation and its relationship to extreme exposure to stress (Binder et al, 2004, 2008; Klengel et al, 2013).
Conclusion
We found that a single exposure to IMO enhances HPA axis activity in mice exposed to FC and EXT, and that HPA alterations may underlie EXT deficits reported here and previously in IMO-exposed mice. Additionally, our data suggest that low-dose dexamethasone suppression of the HPA axis enhanced both within-session EXT learning and EXT RET in a model of the fear-related phenomenology of human PTSD. Additionally, plasma levels of corticosterone and Fkbp5 mRNA expression and DNA methylation in the amygdala were dynamically regulated as a function of both dexamethasone suppression and the fear learning procedures. Together these data suggest that regulation of Fkbp5 mRNA expression via DNA methylation, and its subsequent regulation of GR sensitivity may be implicated in the molecular mechanisms of EXT training and RET.
Funding and Disclosure
The authors declare no conflict of interest.
References
American Psychiatric Association (2013) American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5th edn. American Psychiatric Association: Washington, D.C., xliv, pp 947.
Andero R (2015). Nociceptin and the nociceptin receptor in learning and memory. Prog Neuropsychopharmacol Biol Psychiatry 62: 45–50.
Andero R, Brothers SP, Jovanovic T, Chen YT, Salah-Uddin H, Cameron M et al (2013). Amygdala-dependent fear is regulated by Oprl1 in mice and humans with PTSD. Sci Transl Med 5: 188ra73.
Andero R, Daviu N, Escorihuela RM, Nadal R, Armario A (2012). 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus 22: 399–408.
Andero R, Dias BG, Ressler KJ (2014). A role for Tac2, NkB, and Nk3 receptor in normal and dysregulated fear memory consolidation. Neuron 83: 444–454.
Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ (2011). Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry 168: 163–172.
Appel K, Schwahn C, Mahler J, Schulz A, Spitzer C, Fenske K et al (2011). Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology 36: 1982–1991.
Belda X, Daviu N, Nadal R, Armario A (2012). Acute stress-induced sensitization of the pituitary-adrenal response to heterotypic stressors: independence of glucocorticoid release and activation of CRH1 receptors. Horm Behav 62: 515–524.
Belda X, Fuentes S, Nadal R, Armario A (2008a). A single exposure to immobilization causes long-lasting pituitary-adrenal and behavioral sensitization to mild stressors. Horm Behav 54: 654–661.
Belda X, Rotllant D, Fuentes S, Delgado R, Nadal R, Armario A (2008b). Exposure to severe stressors causes long-lasting dysregulation of resting and stress-induced activation of the hypothalamic-pituitary-adrenal axis. Ann N Y Acad Sci 1148: 165–173.
Binder EB (2009). The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34: S186–S195.
Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB et al (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299: 1291–1305.
Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B et al (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36: 1319–1325.
Buchmann AF, Holz N, Boecker R, Blomeyer D, Rietschel M, Witt SH et al (2014). Moderating role of FKBP5 genotype in the impact of childhood adversity on cortisol stress response during adulthood. Eur Neuropsychopharmacol 24: 837–845.
Cole MA, Kim PJ, Kalman BA, Spencer RL (2000). Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology 25: 151–167.
Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG (2000). Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology 141: 4107–4113.
Ewald ER, Wand GS, Seifuddin F, Yang X, Tamashiro KL, Potash JB et al (2014). Alterations in DNA methylation of Fkbp5 as a determinant of blood-brain correlation of glucocorticoid exposure. Psychoneuroendocrinology 44: 112–122.
Fani N, Gutman D, Tone EB, Almli L, Mercer KB, Davis J et al (2013). FKBP5 and attention bias for threat: associations with hippocampal function and shape. JAMA Psychiatry 70: 392–400.
Fani N, King TZ, Reiser E, Binder EB, Jovanovic T, Bradley B et al (2014). FKBP5 genotype and structural integrity of the posterior cingulum. Neuropsychopharmacology 39: 1206–1213.
Gagliano H, Fuentes S, Nadal R, Armario A (2008). Previous exposure to immobilisation and repeated exposure to a novel environment demonstrate a marked dissociation between behavioral and pituitary-adrenal responses. Behav Brain Res 187: 239–245.
Gavin DP, Chase KA, Sharma RP (2013). Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology 75: 233–245.
Goll MG, Bestor TH (2005). Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481–514.
Gourley SL, Kedves AT, Olausson P, Taylor JR (2009). A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology 34: 707–716.
Hill MN, Tasker JG (2012). Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 204: 5–16.
Ising M, Depping AM, Siebertz A, Lucae S, Unschuld PG, Kloiber S et al (2008). Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur J Neurosci 28: 389–398.
Izquierdo A, Wellman CL, Holmes A (2006). Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci 26: 5733–5738.
Jaenisch R, Bird A (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33: 245–254.
Jiang X, Wang J, Luo T, Li Q (2009). Impaired hypothalamic-pituitary-adrenal axis and its feedback regulation in serotonin transporter knockout mice. Psychoneuroendocrinology 34: 317–331.
Jovanovic T, Norrholm SD, Blanding NQ, Phifer JE, Weiss T, Davis M et al (2010). Fear potentiation is associated with hypothalamic-pituitary-adrenal axis function in PTSD. Psychoneuroendocrinology 35: 846–857.
Jovanovic T, Phifer JE, Sicking K, Weiss T, Norrholm SD, Bradley B et al (2011). Cortisol suppression by dexamethasone reduces exaggerated fear responses in posttraumatic stress disorder. Psychoneuroendocrinology 36: 1540–1552.
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 52: 1048–1060.
Klaassens ER, Giltay EJ, van Veen T, Veen G, Zitman FG (2010). Trauma exposure in relation to basal salivary cortisol and the hormone response to the dexamethasone/CRH test in male railway employees without lifetime psychopathology. Psychoneuroendocrinology 35: 878–886.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Koenen KC, Saxe G, Purcell S, Smoller JW, Bartholomew D, Miller A et al (2005). Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry 10: 1058–1059.
Kohli RM, Zhang Y (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502: 472–479.
Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL et al (2010). Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 151: 4332–4343.
Lee RS, Tamashiro KL, Yang X, Purcell RH, Huo Y, Rongione M et al (2011). A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology (Berl) 218: 303–312.
Maddox SA, Schafe GE, Ressler KJ (2013). Exploring epigenetic regulation of fear memory and biomarkers associated with post-traumatic stress disorder. Front Psychiatry 4: 62.
Maguschak KA, Ressler KJ (2011). Wnt signaling in amygdala-dependent learning and memory. J Neurosci 31: 13057–13067.
Marquez C, Belda X, Armario A (2002). Post-stress recovery of pituitary-adrenal hormones and glucose, but not the response during exposure to the stressor, is a marker of stress intensity in highly stressful situations. Brain Res 926: 181–185.
Mehta D, Binder EB (2012). Gene x environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology 62: 654–662.
Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J et al (2011). Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch Gen Psychiatry 68: 901–910.
Milad MR, Furtak SC, Greenberg JL, Keshaviah A, Im JJ, Falkenstein MJ et al (2013). Deficits in conditioned fear extinction in obsessive-compulsive disorder and neurobiological changes in the fear circuit. JAMA Psychiatry 70: 608–618; quiz 554.
Miller AH, Spencer RL, Pulera M, Kang S, McEwen BS, Stein M (1992). Adrenal steroid receptor activation in rat brain and pituitary following dexamethasone: implications for the dexamethasone suppression test. Biol Psychiatry 32: 850–869.
Moriceau S, Wilson DA, Levine S, Sullivan RM (2006). Dual circuitry for odor-shock conditioning during infancy: corticosterone switches between fear and attraction via amygdala. J Neurosci 26: 6737–6748.
Ninomiya EM, Martynhak BJ, Zanoveli JM, Correia D, da Cunha C, Andreatini R (2010). Spironolactone and low-dose dexamethasone enhance extinction of contextual fear conditioning. Prog Neuropsychopharmacol Biol Psychiatry 34: 1229–1235.
Norrholm SD, Jovanovic T, Olin IW, Sands LA, Karapanou I, Bradley B et al (2011). Fear extinction in traumatized civilians with posttraumatic stress disorder: relation to symptom severity. Biol Psychiatry 69: 556–563.
Ozer EJ, Best SR, Lipsey TL, Weiss DS (2003). Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 129: 52–73.
Paxinos G, Franklin K (2006) The Mouse Brain in Stereotaxic Coordinates. 2nd edn. Academic Press: San Diego.
Pratt WB, Toft DO (1997). Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18: 306–360.
Roozendaal B, McEwen BS, Chattarji S (2009). Stress, memory and the amygdala. Nat Rev Neurosci 10: 423–433.
Sabbagh JJ, O’Leary JC 3rd, Blair LJ, Klengel T, Nordhues BA, Fontaine SN et al (2014). Age-associated epigenetic upregulation of the FKBP5 gene selectively impairs stress resiliency. PLoS One 9: e107241.
Scharf SH, Liebl C, Binder EB, Schmidt MV, Muller MB (2011). Expression and regulation of the Fkbp5 gene in the adult mouse brain. PLoS One 6: e16883.
Smit JW, Weert B, Schinkel AH, Meijer DK (1998). Heterologous expression of various P-glycoproteins in polarized epithelial cells induces directional transport of small (type 1) and bulky (type 2) cationic drugs. J Pharmacol Exp Ther 286: 321–327.
Stroth N, Holighaus Y, Ait-Ali D, Eiden LE (2011). PACAP: a master regulator of neuroendocrine stress circuits and the cellular stress response. Ann N Y Acad Sci 1220: 49–59.
Teicher MH, Samson JA (2013). Childhood maltreatment and psychopathology: a case for ecophenotypic variants as clinically and neurobiologically distinct subtypes. Am J Psychiatry 170: 1114–1133.
VanElzakker MB, Dahlgren MK, Davis FC, Dubois S, Shin LM (2014). From Pavlov to PTSD: the extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem 113: 3–18.
Wessa M, Flor H (2007). Failure of extinction of fear responses in posttraumatic stress disorder: evidence from second-order conditioning. Am J Psychiatry 164: 1684–1692.
Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA et al (2010). Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology 35: 1684–1692.
Yang X, Ewald ER, Huo Y, Tamashiro KL, Salvatori R, Sawa A et al (2012). Glucocorticoid-induced loss of DNA methylation in non-neuronal cells and potential involvement of DNMT1 in epigenetic regulation of Fkbp5. Biochem Biophys Res Commun 420: 570–575.
Yang YL, Chao PK, Lu KT (2006). Systemic and intra-amygdala administration of glucocorticoid agonist and antagonist modulate extinction of conditioned fear. Neuropsychopharmacology 31: 912–924.
Yang YL, Chao PK, Ro LS, Wo YY, Lu KT (2007). Glutamate NMDA receptors within the amygdala participate in the modulatory effect of glucocorticoids on extinction of conditioned fear in rats. Neuropsychopharmacology 32: 1042–1051.
Yehuda R (2009). Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci 1179: 56–69.
Yehuda R, Daskalakis NP, Desarnaud F, Makotkine I, Lehrner AL, Koch E et al (2013). Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front Psychiatry 4: 118.
Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F et al (2014). Lower methylation of glucocorticoid receptor gene promoter 1 in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry 77: 356–364.
Yehuda R, Flory JD, Pratchett LC, Buxbaum J, Ising M, Holsboer F (2010). Putative biological mechanisms for the association between early life adversity and the subsequent development of PTSD. Psychopharmacology (Berl) 212: 405–417.
Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C (2002). The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biol Psychiatry 52: 393–403.
Zannas AS, Binder EB (2014). Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav 13: 25–37.
Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, Uhr M et al (2011). Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry 168: 1107–1116.
Acknowledgements
We would like to thank Dr Kiyoshi Inoue and Dr Nuria Daviu for their assistance in the laboratory. Funding and Disclosure: Support to KJR was provided by the Howard Hughes Medical Institute, NIH (R01MH096764) and by an NIH/NCRR base grant (P51RR000165) to Yerkes National Primate Research Center. TK is supported by a NARSAD Young Investigator Grant (20895) and an EMBO Long-Term Fellowhip (ALTF 1153-2013). RA and KJR were supported by R21MH101492-01 and RA by a NARSAD Young Investigator Grant (22434). AA was supported by Spanish grants from the Ministerio de Economía y Competitividad (SAF2011-28313), Instituto de Salud Carlos III (RD12/0028/0014, Redes Temáticas de Investigación Cooperativa en Salud, Ministerio de Sanidad y Consumo), and Generalitat de Catalunya (SGR2014-1020). TJ has support from R21MH092576 and R01MH100122.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Sawamura, T., Klengel, T., Armario, A. et al. Dexamethasone Treatment Leads to Enhanced Fear Extinction and Dynamic Fkbp5 Regulation in Amygdala. Neuropsychopharmacol 41, 832–846 (2016). https://doi.org/10.1038/npp.2015.210
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.210
This article is cited by
-
Tet Enzyme-Mediated Response in Environmental Stress and Stress-Related Psychiatric Diseases
Molecular Neurobiology (2023)
-
The infralimbic mineralocorticoid blockage prevents the stress-induced impairment of aversive memory extinction in rats
Translational Psychiatry (2022)
-
Sex differences in fear memory consolidation via Tac2 signaling in mice
Nature Communications (2021)
-
Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation
Molecular Psychiatry (2021)
-
Transient receptor potential ankyrin 1 promoter methylation and peripheral pain sensitivity in Crohn’s disease
Clinical Epigenetics (2020)
