
Abstract
The development of posttraumatic stress disorder (PTSD) is influenced by genetic factors. Although there have been some replicated candidates, the identification of risk variants for PTSD has lagged behind genetic research of other psychiatric disorders such as schizophrenia, autism, and bipolar disorder. Psychiatric genetics has moved beyond examination of specific candidate genes in favor of the genome-wide association study (GWAS) strategy of very large numbers of samples, which allows for the discovery of previously unsuspected genes and molecular pathways. The successes of genetic studies of schizophrenia and bipolar disorder have been aided by the formation of a large-scale GWAS consortium: the Psychiatric Genomics Consortium (PGC). In contrast, only a handful of GWAS of PTSD have appeared in the literature to date. Here we describe the formation of a group dedicated to large-scale study of PTSD genetics: the PGC-PTSD. The PGC-PTSD faces challenges related to the contingency on trauma exposure and the large degree of ancestral genetic diversity within and across participating studies. Using the PGC analysis pipeline supplemented by analyses tailored to address these challenges, we anticipate that our first large-scale GWAS of PTSD will comprise over 10 000 cases and 30 000 trauma-exposed controls. Following in the footsteps of our PGC forerunners, this collaboration—of a scope that is unprecedented in the field of traumatic stress—will lead the search for replicable genetic associations and new insights into the biological underpinnings of PTSD.
Similar content being viewed by others

INTRODUCTION
Posttraumatic stress disorder (PTSD) occurs in only a minority of persons exposed to traumatic events (Breslau et al, 1998; Kessler et al, 1995). Factors that influence PTSD susceptibility include sex, age, early life adversity, the nature, and timing of traumatic event exposure(s), the cumulative burden of these exposures, as well as various other psychosocial and personality factors (Zoladz and Diamond, 2013). In the US, race/ethnicity impacts the rate, type, and age at traumatic-event exposure, as well as the risk for development of PTSD after exposure (Roberts et al, 2011). Moreover, some events are more pathogenic than others. Events of an interpersonal nature, eg, rape, partner violence, and assault, confer greater risk of developing PTSD than other types of trauma, eg, natural disasters (Kessler et al, 1995). Twin studies have indicated that risk of exposure to some types of trauma may itself be heritable, which is known as a gene–environment correlation (rGE) effect. Lyons et al. (1993) using data from the Vietnam Era Twin Registry (Eisen et al, 1987; Goldberg et al, 1987), found that the heritability of combat exposure ranged from 35 to 47%. In civilians, Stein et al. (2002) found that exposure to interpersonal traumatic events was modestly heritable (~20%). The rGE for trauma exposure appears to be largely explained by genetic influences on personality (Afifi et al, 2010; Jang et al, 2003). For example, sensation seeking is a heritable personality trait that is characterized by engaging in behavior, such as driving at high speeds (Zuckerman, 1994), which may increase the likelihood of trauma exposure. In addition, the risk of PTSD following trauma exposure is moderately heritable, even after controlling for the genetic influences on trauma exposure. Twin studies established that genetic influences explain a substantial proportion of vulnerability to PTSD, from ~30% in male Vietnam veterans (True et al, 1993), to 38% in a sample of male and female civilians (Stein et al, 2002), with an upward heritability estimate of 72% in young women (Sartor et al, 2011). This is comparable to other internalizing disorders such as major depressive disorder and panic disorder (Kendler and Prescott, 2007). Furthermore, genetic influences on PTSD may overlap with those for other mental disorders. The genetic influences on major depressive disorder and PTSD may substantially overlap (Fu et al, 2007; Koenen et al, 2007; Sartor et al, 2012). Phenotypes like alcohol and drug dependence (Sartor et al, 2011; Xian et al, 2000) and nicotine dependence (Koenen et al, 2005) share ~40% genetic risk with PTSD. Genetic influences common to generalized anxiety disorder and panic disorder symptoms account for ~60% of the genetic variance in PTSD (Chantarujikapong et al, 2001).
The search for genetic markers of PTSD is a relatively new endeavor, with the majority of studies conducted within the last decade. These investigations involve genotyping (measuring variation at) a particular location along the genome. Individuals’ particular genetic code (genotype), at a location (locus) is then compared for a sample of cases and controls. Most research to date has employed the candidate-gene approach, in which genes are selected for study based on their theorized involvement in biological pathways implicated in the pathophysiology of PTSD. Given that PTSD has historically been conceptualized as a disorder of pathological fear and stress (Wilker and Kolassa, 2013), most studies have focused on candidate genes involved in biological systems associated with the fear response, including the hypothalamic–pituitary–adrenal axis (eg, FKBP5, CRHR1) and the locus coeruleus–noradrenergic system (eg, COMT, ADRB1, and ADRB2). Additional work has examined serotonergic and dopaminergic systems involved in the neural pathways underlying emotion (eg, SLC6A4, SLC6A3), and systems involved in memory consolidation and stabilization (eg, WWC1, PRKCA). Candidate gene studies of PTSD have produced a large body of literature (Pitman et al, 2012; Wilker and Kolassa, 2013). However, candidate gene studies have, for the most part, failed to replicate when the definition of replication is restricted to the observation of a significant association in the same allele with the same effect direction (see Sullivan (2007) for a discussion of replication in candidate genes studies). To date, relatively few candidate gene studies of PTSD have examined gene–environment (GxE) interactions, an approach that may be particularly well-suited for examining genetic risk in PTSD. However, candidate gene GxE studies in psychiatric literature have been prone to false positives and suitable replication has been difficult to obtain (Duncan and Keller, 2011). Thus, as in the larger psychiatric genetics literature (Psychiatric Gwas Consortium Coordinating Committee et al, 2009), for the most part, robust support for markers associated with risk or resilience for PTSD has not emerged from candidate gene studies.
In contrast to candidate gene studies, in a genome-wide association study (GWAS), genetic variation—primarily single-nucleotide polymorphisms (SNPs)—is examined without hypothesizing the role of any particular gene or biological function (Psychiatric Gwas Consortium Coordinating Committee et al, 2009). The viability of a GWAS strategy is predicated on the relatively low cost of chip-based genotyping that reliably and cheaply assesses thousands or even millions of SNPs distributed throughout the genome. Chip-based genotyping cannot yield information about rare or even private (present in only one person or shared within a particular family) mutations, except in the case of rare or private large copy number variants (CNVs) that can be detected by examining the assays across multiple SNPs. To examine other types of rare variants, more costly whole-genome or whole-exome sequencing is required. Consequently, the investigation of rare variants has primarily been addressed through sequencing, whereas common-variant associations have been assessed through chip-based genotyping. It is customary to examine hundreds of thousands or millions of SNPs in a single GWAS. As the number of SNPs examined is great, and the number with individually detectable effects is presumably small, strict multiple-testing control is required to reduce the number of false positives. The current customary significance threshold is P<5 × 10−8 for a genome-wide study regardless of the particular number of SNPs examined. This strict threshold is useful in that it gives some assurance that the detected loci will be robustly associated with the disorder under study.
To date, four GWAS of PTSD have been published (Guffanti et al, 2013; Logue et al, 2013; Nievergelt et al, 2015; Xie et al, 2013). The genome-wide significant findings of each are summarized in Table 1. Although the roles of these GWAS-identified genes in risk for PTSD have not been elucidated, the top loci identified in the extant GWAS have been implicated in a variety of processes, including neuroprotection, actin polymerization, neuronal function, and immune function (Almli et al, 2014b; Guffanti et al, 2013; Logue et al, 2013; Xie et al, 2013). Notably, the GWAS have identified variants in novel pathways that would not have been examined using the biologically driven candidate-gene methodology. So far, the findings from the different studies have not consistently implicated a primary set of PTSD risk loci. Numerous factors may contribute to this, including one or more of them being false positives, heterogeneity across samples, and a statistical artifact of the ‘winner’s curse’ which implies that effect size estimates will be inflated for moderately powered studies (Xiao and Boehnke, 2009). It is important to note that samples sizes under 5000 or even 10 000 are now considered to be relatively ‘small’ by modern GWAS standards (Sullivan et al, 2012). Convincing demonstrations of association now come from GWAS of tens or even hundreds of thousands of individuals (Lango Allen et al, 2010).
THE PGC AND PROGRESS IN PSYCHIATRIC GENETICS
Although the results of the PTSD GWAS published to date may prove useful, experience from GWAS of other psychiatric disorders has made it clear that large-scale collaborations are necessary to yield consistently replicable findings. The Psychiatric Genomic Consortium (PGC) was organized in 2007 as an outgrowth of the Genetic Association Information Network—a joint public–private funded venture to study attention deficit/hyperactivity disorder (ADHD), diabetic nephropathy, major depressive disorder, psoriasis, schizophrenia, and bipolar disorder (Gain Collaborative Research Group et al, 2007). The PGC had as its goal to conduct GWAS studies of ADHD, bipolar disorder, major depressive disorder, and schizophrenia, and later autism spectrum disorder (Psychiatric GWAS Consortium Coordinating Committee et al, 2009; The Psychiatric GWAS Consortium Steering Committee, 2009). The PGC was designed to bring together psychiatric GWAS from around the world to enable adequately powered analyses. By centralizing analyses under a consortium umbrella, the PGC has overcome the substantial challenges of harmonizing quality control procedures, analytic methods, and phenotype definitions to enable GWAS meta- and mega-analyses (Sullivan, 2010). By adequately powering analyses, and standing by strict definitions of significance from the outset, the PGC has encouraged the production of high-quality replicable genetic associations.
The PGC has become the largest collaborative effort in the history of psychiatry and, as of this writing, comprises more than 500 scientists from more than 100 countries. More than 172 000 subjects have been included, and genotyping of an additional 90 000 is currently underway. PGC efforts have established that sufficiently powered GWAS is a viable strategy for identifying neuropsychiatric disorder susceptibility loci for bipolar disorder (Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011) and schizophrenia (Ripke et al, 2011). The PGC has enabled discovery of a large number of reliably associated genetic loci, 108 for schizophrenia alone at last count (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). The PGC analyses have also given us an insight into the genetic architecture of psychiatric disorders (Collins and Sullivan, 2013). In particular, these analyses have demonstrated that psychiatric disorders are polygenic (having hundreds or even thousands of risk loci) and that common variation accounts for a substantial component of the underlying genetic architecture. Their results have indicated that GWAS-significant loci represent the tip of the iceberg in terms of the proportion of variance explained by inherited genetic variation, and the remaining variation is likely to represent a mix of rare and common genetic effects. For example, in schizophrenia, the proportion of variation explained by the 108 genome-wide significant loci was 3.4% (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014), whereas estimates of the total proportion of variation explained by common genotyped SNPs has been estimated to be approximately 25 and 45% depending on the population and method used (International Schizophrenia Consortium et al., 2009; Lee et al, 2012; Ripke et al, 2013). In addition to common variants, rare variants such as CNVs were found to explain a proportion of risk for schizophrenia, bipolar disorder, and autism (Malhotra and Sebat, 2012). Results in schizophrenia also suggest that many of the genome-wide significant loci obtained at smaller sample sizes will turn out to be significant once the sample size gets large (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). The polygenic nature of the psychiatric disorders is such that once the sample size is sufficiently large, the genome-wide distribution of association statistics will differ from the expected null distribution (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). A new method called LD regression has been developed to test whether or not genomic inflation in this context represents a polygenic risk component to disease or inflated significance due to the population substructure (Bulik-Sullivan et al, 2015b).
Also importantly, as the list of risk loci has expanded, they have begun to coalesce into biological pathways, illuminating disease pathogenesis and implicating new targets for drug development (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Nurnberger et al, 2014; Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). For example, recent analyses have highlighted the role of immune system and glutamatergic function in schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014) and calcium channel signaling across childhood- and adult-onset disorders (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). The PGC Cross-Disorder Workgroup identified several loci that appear to confer risk across autism, ADHD, bipolar disorder, major depressive disorder, and schizophrenia (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). Aggregate genome-wide analyses (using SNP-heritability estimates and polygene scores) showed significant genetic overlap among these disorders, with the strongest overlap between bipolar disorder and schizophrenia (genetic correlation=+0.68; Cross-Disorder Group of the Psychiatric Genomics Consortium et al, 2013). By highlighting shared biologic vulnerability, this work may inform efforts to refine psychiatric nosology. Recently Bulik-Sullivan et al (2015a) have developed a new computationally efficient technique for estimating cross-trait genetic correlation based on LD regression. The results obtained with this new method mirror earlier work showing genetic correlation between schizophrenia and bipolar disorder. However, this new method has the advantage that it can be run on summary statistics from both traits, rather than necessitating individual-level data.
The PGC has also led the development of the PsychChip. The PsychChip is an Illumina (San Diego, CA) genotyping chip that assesses ~560 000 markers. It is designed to be suitable for analysis of psychiatric traits and for use in the imputation of genome-wide SNP genotypes (described in the Supplementary Materials).
THE PGC-PTSD WORKGROUP
Drs Koenen, Ressler, and Liberzon founded the PCG-PTSD Workgroup (PGC-PTSD) in May 2013 with a satellite meeting at the Society of Biological Psychiatry co-sponsored by NIMH and One Mind, a patient advocacy non-profit organization (http://onemind.org). The PGC-PTSD has, as its goal, the bringing together of a large number of PTSD researchers for the purpose of large-scale GWAS studies of PTSD.
The Sample
The size and characteristics of the groups anticipated to participate in the PGC-PTSD are summarized in Table 2. First, six groups have uploaded genotype data that will be used in the initial PGC-PTSD analysis. This includes a combined sample size of 20 468 subjects (4487 cases and 15 981 controls). Second, an additional 53 552 subjects from 13 studies have been genotyped (19 408 cases and 34 144 controls). Third, there are 20 studies with genotyping in process or planned (N=71 757; 24 439 cases and 47 318 controls). Many of these studies will be using the PsychChip. Data collection sites are from across the US (eg, Atlanta, San Diego, New Haven, Detroit) and include three additional countries (Denmark, The Netherlands and South Africa). Like other PGC disorders, we expect that this initial sample is merely the first foray into large-scale meta-analyses.
The vast majority (>80%) of controls across these studies have experienced a trauma fulfilling the exposure criterion for PTSD. Hence, the PGC-PTSD sample will have a large trauma-exposed control group available for comparison with PTSD cases. Focusing on trauma-exposed individuals may be useful, as any PTSD risk allele, which will have an increased rate in PTSD cases, will presumably have a lower frequency in PTSD-negative trauma-exposed controls than in trauma negative or unscreened controls. Hence, all other things being equal, the greater difference in allele frequency between PTSD cases and trauma-exposed controls will lead to a greater power to detect the associations than a sample that includes trauma-negative or unscreened controls. Utilizing unscreened controls in the presence of rGE effects could result in associations representing a mix of loci, some of which were associated with risk of PTSD in the presence of trauma exposure and some of which were related to the risk of trauma exposure itself. The use of only trauma-exposed controls and inclusion of degree of trauma exposure as a covariate in analyses should be adequate to place our focus tightly on loci that increase risk of PTSD directly.
Phenotype and Exposure Measurement Complexity
The harmonization of data across PGC–PTSD groups, like that for other psychiatric disorders, is complicated by variability in the assessment methods used. Two major approaches to the assessment of PTSD symptoms and diagnosis—structured clinical interviews and self-report instruments—are represented, with the primary distinction between them related to the source of the data (ie, clinician ratings vs participant self-report). Of the six samples already uploaded to the PGC–PTSD, five used self-report measures and one used clinician ratings. The major limitation of clinical interviews is the considerable time and expense involved in training and administration, which renders this approach impractical for many studies. Studies featuring some of the largest samples have used self-report instruments to assess symptoms and estimate diagnosis. Additionally, although not yet investigated within the PGC–PTSD group, there is the possibility of using diagnostic information from additional sources such as from electronic health records, which can provide evidence of convergent validity. Finally, methods for determining diagnostic status (ie, cases vs controls) differ between interview and self-report approaches, as well as across traumatized populations. Interview-based studies, based on the judgment of trained clinicians, typically apply the DSM algorithm (ie, for DSM-IV, one reexperiencing symptom, three avoidance and numbing symptoms, and two hyperarousal symptoms). With self-report measures, diagnostic classifications are somewhat less straightforward. The DSM-IV algorithm can also be applied by defining symptoms endorsed above a given severity threshold level as present (ie, causing moderate or greater distress). However, patterns of item endorsement tend to vary across items and populations, so the application of a uniform criterion can yield significant differences in composition of cases across samples. Alternatively, PTSD diagnostic status can be determined in relation to a total symptom severity score cutoff. Studies that have examined the relationship between probable diagnoses derived from this approach vs interview measures of PTSD have found acceptable, though not excellent, agreement (see eg, McDonald et al, 2009; McDonald and Calhoun, 2010). Studies have shown that for any given measure the optimal score for differentiating cases from controls differs across samples and can be influenced by a host of factors, most notably, the base rate of the diagnosis in the sample (for a meta-analysis of PTSD Checklist (PCL) studies, see Terhakopian et al, 2008). Thus, because the same instrument can yield different classification performance across different samples, our cutoff score selections will take into account independent estimates of the true base rate of the sample.
The harmonization of measures of trauma exposure across studies is an additional complication for PTSD genetics research. Though the DSM offers a broad definition of the types of events known to cause PTSD, there is no uniform or generally agreed-upon framework for categorizing or measuring them. A variety of self-report measures of trauma exposure are represented among PGC–PTSD studies. Most consist of a list of events that meet the DSM-IV PTSD Criterion A1 trauma definition including exposure to sexual or physical assault, combat or warfare, sudden death of friend/loved one, and so on. Most also make it possible to re-classify events for harmonization purposes into broader categories such as childhood vs adult trauma, or interpersonal versus non-interpersonal trauma, or to compute a measure of total trauma load (ie, a sum of event exposure categories across the lifespan).
The instruments used in the various studies also differ with respect to the way that they link PTSD to the trauma. In clinical interview instruments such as the Clinician Administered PTSD Scale for DSM-IV (Blake et al, 1990) and the Structured Clinical Interview for DSM-IV Axis I Disorders (First et al, 1994), interviewers identify an index event(s) and then evaluate its link to subsequent symptoms while accounting for confounding factors such as comorbidity, substance abuse, medical issues, and reporting style. For self-report measures (eg, the PCL; Weathers et al, 1993), approaches range from those that link symptoms to a single event, to those that do not reference a single event, to those that reference military experience broadly.
Ancestry
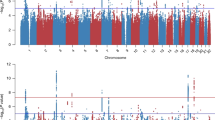
Most extant PGC GWAS have been restricted to a single ancestral population because population stratification can lead to Type I and Type II errors in genetic association studies (Marchini et al, 2004). Psychiatric research in the US and Europe has traditionally enrolled a relatively large proportion of subjects of European ancestry, and consequently, GWAS in the PGC have been performed primarily using subjects of European ancestry (Figure 1a). In contrast, PTSD studies have recruited subjects primarily from high-risk populations, such as combat-veteran cohorts, or in urban areas with high rates of violent crime, and thus PGC–PTSD samples include a large proportion of subjects of African-American and Hispanic/Latino ancestry (Figure 1b). GWAS on such heterogeneous and admixed samples require additional considerations (eg, a study by Pasaniuc et al (2011)). Combining across multiple ancestry groups via meta-analysis has become more common in the recent past (see eg, Nievergelt et al, 2015 and Li and Keating, 2014 for review).

A comparison of ancestral diversity in (a) representative Psychiatric Genomics Consortium (PGC) samples of primarily European ancestry and (b) representative PGC–PTSD samples. Key: mrsa, mrsb—subsets (a and b) of the Marine Resilience Study (Nievergelt); gtpx—Grady Trauma Project (Ressler); gsdx—Genetics of Substance Dependence (Gelernter); fscd—Family Studies of Cocaine Dependence (Bierut); dnhs—Detroit Neighborhood Health Study (Aiello); cogb, coga—subsets (a and b) of the COGEND study (Bierut); Note that African American refers to subjects from the USA who typically have a mix of African and European ancestry, whereas African Ancestry refers to subjects from Africa without admixed ancestry. PTSD, posttraumatic stress disorder.
RESEARCH STRATEGY
PTSD Meta-Analysis
Our proposed analysis strategy is described in the Supplementary Materials and is briefly outlined here. Standardized quality control procedures and GWAS analysis based on the PGC GWAS analysis pipeline will be used (Ripke et al, 2013). Harmonized versions of continuous predictive variables and outcomes (eg, PTSD severity) will be generated based on within-study normalization of the instruments available. Categorical outcome and predictor variables will, for the most part, be based on the diagnostic schema adopted by the principal investigator of the particular study taking into account sample and measurement factors that affect prevalence estimates. The efficacy of the harmonization will be assessed using the descriptive statistics and by examining correlations between predictive variables, outcomes, and reported demographic information from each group. Our primary analysis will be a GWAS meta-analysis of PTSD followed by a GWAS of PTSD severity, both controlling for potential sources of bias as well as trauma-exposure variables. Based on a consensus of participating group members at in-person PGC–PTSD planning meetings, we determined to utilize dichotomous DSM-IV diagnosis as the primary phenotype. Initially, this analysis will be restricted to trauma-exposed controls. The pipeline will be modified to account for greater population stratification between and within PGC–PTSD groups compared with the typical PGC analysis. Both within-ancestral group and cross-ancestral group meta-analysis will be performed. Subsequent investigation will include analyses of rare variants, including structural variants such as CNV.
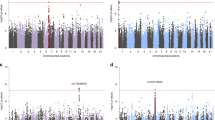
The PGC–PTSD has already assembled a substantial aggregate sample size, as well as an extensive set of samples that will be genotyped if funding allows. The power to detect a SNP effect in a GWAS analysis varies as a function of the size of the effect, the allele frequency of the SNP, and the sample size. This is illustrated in Figure 2, which displays the minimum effect size that yields 80% power as a function of the SNP allele frequency and sample size. An analysis including the 45 000, the samples that have currently funded genotyping will have 80% power to detect a locus with a genotype relative risk (GRR) between 1.2 and 1.11 for allele frequencies between 5 and 20%. Increasing the sample size to 60 000 will allow us to detect a locus with a GRR between 1.17 and 1.1, respectively.

Effect size necessary to have 80% power for case-control and quantitative-trait association analyses demonstrating the relation between increasing sample size and ability to detect loci of smaller effect sizes. Key: calculated assuming PTSD prevalence of 15%, additive model, a type I error rate of 5 × 10−8, and perfect LD between marker and trait allele for MAF>5%). Calculations were based on a 1 : 3 PTSD case-control ratio or quantitative traits such as PTSD symptoms. PTSD, posttraumatic stress disorder.
GxE Analyses
In addition to the standard GWAS meta-analysis, a secondary aim of the PGC–PTSD is to conduct a series of GxE analyses. Some readers may be surprised that this is not the primary analysis for PTSD. Although we are well aware of the conceptual relevance of GxE models to PTSD, the statistical challenges associated with GxE analyses are formidable. First, although PTSD clearly results from the interaction of trauma with genetic predisposition, it is unclear whether or not the biological realities of such an interaction are captured by testing deviations from a multiplicative logistic regression model (Thompson, 1991). Second, the significance of the GxE interaction term estimated using standard regression models can be inflated under commonly occurring conditions (Almli et al, 2014a; Voorman et al, 2011). Third, obtaining reasonable power in GxE analysis takes sample sizes larger than those required for main effect analyses. A sample four times as large has been proposed as a rule of thumb (Thomas, 2010). Finally, the power and bias of different GxE analysis methods vary depending on the nature of the interaction (Cornelis et al, 2012; Mukherjee et al, 2012).
Approaches used previously in PTSD genetics studies have ranged from including cumulative lifetime ‘trauma load’ as a covariate in the analysis (Kolassa et al, 2010) to explicitly testing for GxE interactions (Digangi et al, 2013). To date, there have been no genome-wide GxE studies of PTSD. Although the single genome-wide GxE study published in psychiatry to date (a study of ADHD) did not yield significant findings (Sonuga-Barke et al, 2008), genome-wide GxE studies have been successful in other areas (eg, Beaty et al, 2011).
The PGC–PTSD will use a two-stage strategy to examine GxE effects. First, given the likelihood of developing PTSD increases with exposure to childhood trauma, interpersonal violence, and with increasing trauma load, GxE models will test the hypothesis that the effects of risk variants for PTSD (identified through the primary GWAS) are moderated by these environmental variables. The second approach is a ‘genome-wide GxE’ meta-analysis approach that will systematically interrogate the genome for GxE effects between SNPs and these three environmental variables. This will include fitting a logistic regression model of PTSD and linear model of PTSD severity as a function of a SNP × childhood trauma, SNP × interpersonal trauma, and SNP × total trauma load interaction effects using robust SEs to combat genome-wide inflation of significance. Follow-up analyses will examine the effect of multiple characteristics of trauma exposure, including trauma load, type, timing, and severity. Finally, we note that the data gathered here will provide a resource for secondary analysis and methodological development, as has been the case for other PGC disorders such as schizophrenia.
Comorbidity
PTSD is highly comorbid with other psychiatric disorders, and a substantial proportion of this comorbidity may be explained by common genetic influences (Koenen et al, 2003; Wolf et al, 2010). Hence, in this context comorbidity may present an opportunity to explore potential overlapping genetic effects in our sample. We propose to follow the PGC cross-disorder model and perform a polygenic architecture analysis with polygenic risk scores and LD regression to determine the proportion of genetic variance (heritability) common across PTSD and other psychiatric disorders.
PGC–PTSD SUBGROUPS
The PGC–PTSD also represents the confluence of vast reserves of PTSD-related information that will enable large-sample investigations of PTSD-associated epigenetic, neuroimaging, and other neurobiological measures. In order to facilitate the analysis of these data, a pair of focus groups have been created within the PGC–PTSD workgroup: the PGC–PTSD Epigenetics Workgroup and the PGC–PTSD Neuroimaging Genetics Workgroup.
PGC–PTSD Epigenetics Workgroup
Recently, ‘stand alone’ genome-scale studies of PTSD epigenetics and gene expression have provided initial insight into molecular dysregulation associated with the disorder (Mehta et al, 2013). Epigenetics provides a molecular context to GxE interactions by offering a biological mechanism through which gene expression can vary in response to an environmental exposure (see eg, Latham et al, 2012). Genetic variation has been shown to influence DNA methylation and gene expression levels, often in tissue-specific and developmental stage-specific manners; so-called methylation trait quantitative loci (meQTLs) and expression trait quantitative loci (eQTLs) have been identified across the genome in numerous studies to date (see eg, Smith et al, 2014). Although genome-scale studies of PTSD-associated meQTLs and eQTLs have yet to be reported, focused candidate gene studies have revealed notable examples of each (see eg, Klengel et al, 2013; Mehta et al, 2011; Ressler et al, 2011). Within the PGC–PTSD, there are several groups with both genome-wide genotype and methylation data, with a current total n=1114. The PGC–PTSD Methylation Workgroup has, as its goal, the creation of a large PTSD-focused methylation data set that can be used to identify whether gene expression or methylation act as mediators of the association between SNPs and PTSD risk as well as identifying PTSD-relevant eQTLs and meQTLs that can be examined for association to PTSD and trauma exposure.
PGC–PTSD Neuroimaging Workgroup
The PGC group members have a large number of participating groups with neuroimaging data. Within the PGC–PTSD there are over 5000 samples that will have both structural MRI and GWAS data available. Smaller data sets of DTI, resting-state fMRI, MEG, and other imaging modalities are also available. These data will allow the investigation of how genomic markers modulate neuroimaging quantitative traits (QTs) associated with PTSD. The uncertainty associated with psychiatric nosology makes reference to an intermediate biological variable attractive, as the heritability of intermediate phenotypes such as regional brain volumes is often 80% or higher (den Braber et al, 2013). However, these will not represent a magic bullet. Given the results of the ENIGMA group, a neuroimaging GWAS meta-analysis consortium, effect sizes observed for individual SNPs on brain structures are likely to be modest and require large sample sizes to be adequately powered (Hibar et al, 2015; Stein et al, 2012). The PGC–PTSD Neuroimaging Workgroup will facilitate the creation of a large PTSD-focused neuroimaging data set to investigate genomic markers for association to cortical and subcortical volumes such as hippocampus, amygdala, and medial prefrontal cortex structures as well as regional cortical thickness changes that are associated with PTSD. Genomic markers found to predict imaging QTs may have a role in PTSD symptoms or diagnoses (Meyer-Lindenberg and Weinberger, 2006).
DISCUSSION
There are several ways in which the PGC–PTSD will represent and advance the current cutting-edge of PTSD genetics research. First and foremost, the PGC–PTSD will build on what the PGC has learned in other disease domains. We believe that the PGC–PTSD, through its investigation of genetic variation, epigenetic variation, and neuroimaging characteristics of PTSD will provide new and important insights into the biological underpinnings of PTSD risk. The PGC–PTSD additionally has the goal of developing clinically useful biomarkers of PTSD. The work of the PGC–PTSD will inform the development of at least three types of clinical biomarkers. The first are predictive biomarkers that reliably distinguish persons at high vs low risk for the development of PTSD following trauma. A gene or combination of genes associated with PTSD may, in conjunction with other information, contribute to an algorithm for estimating the risk of developing PTSD. Such a risk algorithm could be used in first-response settings or the military to better target preventive interventions.
The second type of biomarker likely to be informed by the discoveries of the PGC PTSD working group is related to treatment matching. There are several effective interventions for PTSD including prolonged exposure, cognitive processing therapy, skills training in affective and interpersonal regulation, and pharmacological interventions. However, little is known about which of these treatments might be most effective for which patients. One of the long-term goals of the PGC–PTSD will be to examine whether patients with specific combinations of genetic variants and environmental exposures respond differentially to evidence-based treatments.
The third type of biomarker that may be informed by the work of the PGC–PTSD is relapse prediction. Several of the studies included in the PGC–PTSD meta-analysis are longitudinal and a few are truly prospective (Baker et al, 2012; Goldmann et al, 2011). Thus, we will be able to examine whether genetic variants associated with PTSD also predict the clinical course of the disorder. If patients with a specific combination of genetic and environmental risk factors are at higher risk of relapse, such patients could be targeted with relapse prevention strategies.
Knowledge of the genetic and environmental architecture of PTSD has the potential to advance our understanding of the pathophysiology of the disorder and inform treatment development. Of particular interest is the development of preventive pharmacological interventions for PTSD that could be administered in the acute aftermath of traumatic events. Many pharmacological agents have been explored in this regard including propranolol and hydrocortisone, but none have shown decisive efficacy for PTSD prevention in large RCTs. The success of GWAS of schizophrenia and bipolar disorder has led to the identification of new treatment targets (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Nurnberger et al, 2014; Schizophrenia Working Group of the Psychiatric Genomics Consortium 2014). Clinical trials are underway to determine whether these will translate into more effective treatments. Rather than simply generating a list of associated DNA variants, our goal is to produce the same successes for PTSD.
FUNDING AND DISCLOSURE
This study is supported by One Mind. Dr Morey was supported by Department of Veterans Affairs (DVA) 1I01CX000748-01A1 and I01CX000120-01 and the Mid-Atlantic Mental Illness, Research, and Education Center. MBS:Consultant: Care Management Technologies; Janssen, Pfizer, and Tonix Pharmaceuticals Editorial Work: Up-To-Date; Biological Psychiatry (journal); Depression and Anxiety (journal). Dr KJR currently receives funding from NIH (NIMH) and HHMI. In the past, he has received funding from the Burroughs Wellcome Fund, and the Brain and Behavior Fund. He is a founding member of Extinction Pharmaceuticals/Therapade Technologies to develop D-cycloserine, a generically available compound, for use to augment the effectiveness of psychotherapy. He has received no equity or income from this relationship within the last 5 years. JWS has received funding from ProMedica Klarman Family Foundation ANGI Welcome Trust Strategic Awards Committee (London). In the past 3 years, MWL has received funding from the US Department of Veterans Affairs, the NIH, and the Thome Memorial Foundation. Dr AKS receives or has received research support from the American Foundation for Suicide Prevention, Schering Plough Pharmaceuticals, NARSAD (YI 19233), the Conquer Cancer Foundation and NIH (MH085806, MH088609, DK100392 and MD009064). The remaining authors declare no conflict of interest. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
References
Afifi TO, Asmundson GJ, Taylor S, Jang KL (2010). The role of genes and environment on trauma exposure and posttraumatic stress disorder symptoms: a review of twin studies. Clin Psychol Rev 30: 101–112.
Almli LM, Duncan R, Feng H, Ghosh D, Binder EB, Bradley B et al (2014a). Correcting systematic inflation in genetic association tests that consider interaction effects: application to a genome-wide association study of posttraumatic stress disorder. JAMA Psychiatry 71: 1392–1399.
Almli LM, Srivastava A, Fani N, Kerley K, Mercer KB, Feng H et al (2014b). Follow-up and extension of a prior genome-wide association study of posttraumatic stress disorder: gene x environment associations and structural magnetic resonance imaging in a highly traumatized African-American civilian population. Biol Psychiatry 76: e3–e4.
Baker DG, Nash WP, Litz BT, Geyer MA, Risbrough VB, Nievergelt CM et al (2012). Predictors of risk and resilience for posttraumatic stress disorder among ground combat Marines: methods of the Marine Resiliency Study. Prev Chronic Dis 9: E97.
Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB et al (2011). Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol 35: 469–478.
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Charney DS, Keane TM (1990) The Clinician-Administered PTSD Scale-IV. National Center for Posttraumatic Stress Disorder, Behavioral Sciences Division: Boston.
Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P (1998). Trauma and posttraumatic stress disorder in the community: the 1996 Detroit area survey of trauma. Arch Gen Psychiatry 55: 626–632.
Bulik-Sullivan BK, Finucane HK, Anttila V, Gusev A, Day FR et alReprogen Consortium (2015a). An atlas of genetic Correlations across human diseases and traits. BioRxiv http://dx.doi.org/10.1101/014498.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J et alSchizophrenia Working Group of the Psychiatric Genomics C (2015b). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295.
Chantarujikapong SI, Scherrer JF, Xian H, Eisen SA, Lyons MJ, Goldberg J et al (2001). A twin study of generalized anxiety disorder symptoms, panic disorder symptoms and post-traumatic stress disorder in men. Psychiatry Res 103: 133–145.
Collins AL, Sullivan PF (2013). Genome-wide association studies in psychiatry: what have we learned? Br J Psychiatry 202: 1–4.
Cornelis MC, Tchetgen EJ, Liang L, Qi L, Chatterjee N, Hu FB et al (2012). Gene-environment interactions in genome-wide association studies: a comparative study of tests applied to empirical studies of type 2 diabetes. Am J Epidemiol 175: 191–202.
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381: 1371–1379.
Cross-Disorder Group of the Psychiatric Genomics Consortium Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al (2013). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45: 984–994.
den Braber A, Bohlken MM, Brouwer RM, van 't Ent D, Kanai R, Kahn RS et al (2013). Heritability of subcortical brain measures: a perspective for future genome-wide association studies. Neuroimage 83: 98–102.
Digangi J, Guffanti G, McLaughlin KA, Koenen KC (2013). Considering trauma exposure in the context of genetics studies of posttraumatic stress disorder: a systematic review. Biol Mood Anxiety Disord 3: 2.
Duncan LE, Keller MC (2011). A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am J Psychiatry 168: 1041–1049.
Eisen S, True WR, Goldberg J, Henderson W, Robinette CD (1987). The Vietnam Era Twin Registry: method of construction. Acta Genet Med Gemellol 36: 61–66.
First MB, Spitzer RL, Gibbon M, Williams J (1994) Structured Clinical Interview for Axis I DSM-IV Disorders. Biometrics Research: New York.
Fu Q, Koenen KC, Miller MW, Heath AC, Bucholz KK, Lyons MJ et al (2007). Differential etiology of posttraumatic stress disorder with conduct disorder and major depression in male veterans. Biol Psychiatry 62: 1088–1094.
Gain Collaborative Research Group, Manolio TA, Rodriguez LL, Brooks L, Abecasis G et alCollaborative Association Study of P et al (2007). New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet 39: 1045–1051.
Goldberg J, True WR, Eisen SA, Henderson WG, Robinette C.D. (1987). The Vietnam Era Twin (VET) Registry: ascertainment bias. Acta Ganet Med Gemellol 36: 67–78.
Goldmann E, Aiello A, Uddin M, Delva J, Koenen K, Gant LM et al (2011). Pervasive exposure to violence and posttraumatic stress disorder in a predominantly African American Urban Community: the Detroit Neighborhood Health Study. J Trauma Stress 24: 747–751.
Guffanti G, Galea S, Yan L, Roberts AL, Solovieff N, Aiello AE et al (2013). Genome-wide association study implicates a novel RNA gene, the lincRNA AC068718.1, as a risk factor for post-traumatic stress disorder in women. Psychoneuroendocrinology 38: 3029–3038.
Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N et al (2015). Common genetic variants influence human subcortical brain structures. Nature 520: 224–229.
International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC et al (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460: 748–752.
Jang KL, Stein MB, Taylor S, Asmundson GJ, Livesley WJ (2003). Exposure to traumatic events and experiences: aetiological relationships with personality function. Psychiatry Res 120: 61–69.
Kendler KS, Prescott CA (2007) Genes, Environment, and Psychopathology: Understanding the Causes of Psychiatric and Substance Use Disorders. The Guilford Press: New York, NY.
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 52: 1048–1060.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Koenen KC, Fu QJ, Ertel K, Lyons MJ, Eisen SA, True WR et al (2007). Common genetic liability to major depression and posttraumatic stress disorder in men. J Affect Disord 105: 109–115.
Koenen KC, Hitsman B, Lyons MJ, Niaura R, McCaffery J, Goldberg J et al (2005). A twin registry study of the relationship between posttraumatic stress disorder and nicotine dependence in men. Arch Gen Psychiatry 62: 1258–1265.
Koenen KC, Lyons MJ, Goldberg J, Simpson J, Williams WM, Toomey R et al (2003). A high risk twin study of combat-related PTSD comorbidity. Twin Res 6: 218–226.
Kolassa IT, Kolassa S, Ertl V, Papassotiropoulos A, De Quervain DJ (2010). The risk of posttraumatic stress disorder after trauma depends on traumatic load and the catechol-o-methyltransferase Val(158)Met polymorphism. Biol Psychiatry 67: 304–308.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F et al (2010). Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467: 832–838.
Latham KE, Sapienza C, Engel N (2012). The epigenetic lorax: gene-environment interactions in human health. Epigenomics 4: 383–402.
Lee SH, DeCandia TR, Ripke S, Yang J et alSchizophrenia Psychiatric Genome-Wide Association Study C, International Schizophrenia C (2012). Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 44: 247–250.
Li YR, Keating BJ (2014). Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med 6: 91.
Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, Reardon AF et al (2013). A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol Psychiatry 18: 937–942.
Lyons MJ, Goldberg J, Eisen SA, True W, Tsuang MT, Meyer JM et al (1993). Do genes influence exposure to trauma? A twin study of combat. Am J Med Genet 48: 22–27.
Malhotra D, Sebat J (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148: 1223–1241.
Marchini J, Cardon LR, Phillips MS, Donnelly P (2004). The effects of human population structure on large genetic association studies. Nat Genet 36: 512–517.
McDonald SD, Beckham JC, Morey RA, Calhoun PS (2009). The validity and diagnostic efficiency of the Davidson Trauma Scale in military veterans who have served since September 11th, 2001. J Anxiety Disord 23: 247–255.
McDonald SD, Calhoun PS (2010). The diagnostic accuracy of the PTSD checklist: a critical review. Clin Psychol Rev 30: 976–987.
Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J et al (2011). Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch Gen Psychiatry 68: 901–910.
Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW et al (2013). Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA 110: 8302–8307.
Meyer-Lindenberg A, Weinberger DR (2006). Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci 7: 818–827.
Mukherjee B, Ahn J, Gruber SB, Chatterjee N (2012). Testing gene-environment interaction in large-scale case-control association studies: possible choices and comparisons. Am J Epidemiol 175: 177–190.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW et al (2015). Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: a genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology 51: 459–471.
Nurnberger JI Jr ., Koller DL, Jung J, Edenberg HJ, Foroud T, Guella I et al (2014). Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry 71: 657–664.
Pasaniuc B, Zaitlen N, Lettre G, Chen GK, Tandon A, Kao WH et al (2011). Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet 7: e1001371.
Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW et al (2012). Biological studies of post-traumatic stress disorder. Nat Rev Neurosci 13: 769–787.
Psychiatric GWAS Consortium Bipolar Disorder Working Group (2011). Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43: 977–983.
Psychiatric GWAS Consortium Coordinating Committee, Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV et al (2009). Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 166: 540–556.
Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K et al (2011). Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470: 492–497.
Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S et al (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 45: 1150–1159.
Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA et al (2011). Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43: 969–976.
Roberts AL, Gilman SE, Breslau J, Breslau N, Koenen KC (2011). Race/ethnic differences in exposure to traumatic events, development of post-traumatic stress disorder, and treatment-seeking for post-traumatic stress disorder in the United States. Psychol Med 41: 71–83.
Sartor CE, Grant JD, Lynskey MT, McCutcheon VV, Waldron M, Statham DJ et al (2012). Common heritable contributions to low-risk trauma, high-risk trauma, posttraumatic stress disorder, and major depression. Arch Gen Psychiatry 69: 293–299.
Sartor CE, McCutcheon VV, Pommer NE, Nelson EC, Grant JD, Duncan AE et al (2011). Common genetic and environmental contributions to post-traumatic stress disorder and alcohol dependence in young women. Psychol Med 41: 1497–1505.
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511: 421–427.
Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ et al (2014). Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics 15: 145.
Sonuga-Barke EJ, Lasky-Su J, Neale BM, Oades R, Chen W, Franke B et al (2008). Does parental expressed emotion moderate genetic effects in ADHD? An exploration using a genome wide association scan. Am J Med Genet B Neuropsychiatr Genet 147B: 1359–1368.
Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM et al (2012). Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet 44: 552–561.
Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ (2002). Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: a twin study. Am J Psychiatry 159: 1675–1681.
Sullivan PF (2007). Spurious genetic associations. Biol Psychiatry 61: 1121–1126.
Sullivan PF (2010). The psychiatric GWAS consortium: big science comes to psychiatry. Neuron 68: 182–186.
Sullivan PF, Daly MJ, O'Donovan M (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 13: 537–551.
Terhakopian A, Sinaii N, Engel CC, Schnurr PP, Hoge CW (2008). Estimating population prevalence of posttraumatic stress disorder: an example using the PTSD checklist. J Trauma Stress 21: 290–300.
The Psychiatric GWAS Consortium Steering Committee (2009). A framework for interpreting genome-wide association studies of psychiatric disorders. Mol Psychiatry 14: 10–17.
Thomas D (2010). Gene–environment-wide association studies: emerging approaches. Nat Rev Genet 11: 259–272.
Thompson WD (1991). Effect modification and the limits of biological inference from epidemiologic data. J Clin Epidemiol 44: 221–232.
True WR, Rice J, Eisen SA, Heath AC, Goldberg J, Lyons MJ et al (1993). A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch Gen Psychiatry 50: 257–264.
Voorman A, Lumley T, McKnight B, Rice K (2011). Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS One 6: e19416.
Weathers FW, Litz BT, Herman DS, Huska JA, Keane TM (1993). The PTSD checklist: Reliability, validity, & diagnostic utility. Annual Meeting of the International Society for Traumatic Stress Studies,: San Antonio, TX.
Wilker S, Kolassa IT (2013). The formation of a neural fear network in posttraumatic stress disorder: insights from molecular genetics. Clin Psychol Sci 1: 452–469.
Wolf EJ, Miller MW, Krueger RF, Lyons MJ, Tsuang MT, Koenen KC (2010). Posttraumatic stress disorder and the genetic structure of comorbidity. J Abnorm Psychol 119: 320–330.
Xian H, Chantarujikapong SI, Scherrer JF, Eisen SA, Lyons MJ, Goldberg J et al (2000). Genetic and environmental influences on posttraumatic stress disorder, alcohol and drug dependence in twin pairs. Drug Alcohol Depend 61: 95–102.
Xiao R, Boehnke M (2009). Quantifying and correcting for the winner's curse in genetic association studies. Genet Epidemiol 33: 453–462.
Xie P, Kranzler HR, Yang C, Zhao H, Farrer LA, Gelernter J (2013). Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol Psychiatry 74: 656–663.
Zoladz PR, Diamond DM (2013). Current status on behavioral and biological markers of PTSD: a search for clarity in a conflicting literature. Neurosci Biobehav Rev 37: 860–895.
Zuckerman M (1994) Behavioral Expressions and Biosocial Bases of Sensation Seeking. Cambridge University Press: New York, NY.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
PowerPoint slides
Rights and permissions
About this article

Cite this article
Logue, M., Amstadter, A., Baker, D. et al. The Psychiatric Genomics Consortium Posttraumatic Stress Disorder Workgroup: Posttraumatic Stress Disorder Enters the Age of Large-Scale Genomic Collaboration. Neuropsychopharmacol 40, 2287–2297 (2015). https://doi.org/10.1038/npp.2015.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.118
This article is cited by
-
Post-traumatic stress disorder: clinical and translational neuroscience from cells to circuits
Nature Reviews Neurology (2022)
-
Rare copy number variation in posttraumatic stress disorder
Molecular Psychiatry (2022)
-
Neurobiological consequences of racial disparities and environmental risks: a critical gap in understanding psychiatric disorders
Neuropsychopharmacology (2020)
-
Effects of COMT rs4680 and BDNF rs6265 polymorphisms on brain degree centrality in Han Chinese adults who lost their only child
Translational Psychiatry (2020)
-
FKBP5 haplotypes and PTSD modulate the resting-state brain activity in Han Chinese adults who lost their only child
Translational Psychiatry (2020)
