
Abstract
As acute ethanol exposure inhibits N-methyl-D-aspartate glutamate (Glu) receptors, sudden withdrawal from chronic alcohol use may lead to an increased activation of these receptors with excitotoxic effects. In the longer term, brain levels of Glu and its metabolites, such as glutamine (Gln), are likely to be chronically altered by alcohol, possibly providing a measure of overall abnormal Glu–Gln cycling. However, few studies have assessed concentrations of these metabolites in clinical populations of individuals with alcohol use disorders. Glu and Gln levels were compared in groups of 17 healthy controls and in 13 participants with alcohol dependence. Within the alcohol-dependent group, seven participants had current alcohol use disorder (AUD), and six had AUD in remission for at least 1 year (AUD-R). Neurometabolite concentrations were measured with proton magnetic resonance spectroscopy (1H-MRS) in a predominantly gray matter voxel that included the bilateral anterior cingulate gyri. Tissue segmentation provided an assessment of the proportion of gray matter in the 1H-MRS voxel. The Drinker Inventory of Consequences (DrInC) and Form-90 were administered to all participants to quantify alcohol consequences and use. Glu level was lower and Gln level was higher in the AUD and AUD-R groups relative to the control group; creatine, choline, myo-inositol, and total N-acetyl groups, primarily N-acetylaspartate did not differ across groups. These results were not confounded by age, sex, or proportion of gray matter in the 1H-MRS voxel. Neurometabolite concentrations did not differ between AUD and AUD-R groups. Subsequent regressions in the combined clinical group, treating voxel gray matter proportion as a covariate, revealed that total score on the DrInC was positively correlated with Gln but negatively correlated with both Glu and gray matter proportion. Regression analyses, including DrInC scores and smoking variables, identified a marginal independent effect of smoking on Gln. The current findings of higher Gln and lower Glu in the combined AUD and AUD-R groups might indicate a perturbation of the Glu–Gln cycle in alcohol use disorders. The absence of differences in mean Glu and Gln between the AUD and AUD-R groups suggests that altered Glu–Gln metabolism may either predate the onset of abuse or persist during prolonged abstinence.
Similar content being viewed by others
INTRODUCTION
Alcohol is one of the most commonly used recreational drugs in society today, as almost half of Americans over the age of 12 years (48.3%) report being current drinkers (US Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, 2002). Results from the 2001 National Household Survey on Drug Abuse showed that approximately one-fifth of Americans over the age of 12 years reported binge drinking at least once in the last 30 days (ibid). In addition, roughly 14% of the US population has met criteria for alcohol dependence at some point, with 7% meeting criteria in the previous year (APA, 1994). It has been estimated that approximately 14% of those with alcohol use disorders have persistent cognitive impairment (Rourke and Loberg, 1996).
Glutamate (Glu) metabolism abnormality is characteristic of psychopathological disorder (Bustillo et al, 2011) and traumatic brain injury (Yeo et al, 2011), and glutamatergic neurotoxicity has been implicated as a major component of the neurodegenerative effects of alcohol use disorders. Acute exposure to alcohol inhibits ionotropic Glu receptors, reducing the overall excitatory synaptic transmission (Lovinger, 1993). This effect is strongest at the N-methyl-D-aspartate receptor (NMDAR), though there is also evidence for involvement of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate- and kainate-type Glu receptors in certain brain regions (Lovinger, 1993; Frye and Fincher, 2000). Chronic ethanol-induced inhibition leads to a compensatory upregulation of NMDAR in cortical neurons (Hoffman, 1995). This upregulation is thought to result in hyperexcitability during alcohol withdrawal and, hence, an increase in synaptic Glu concentrations in cortical regions (Rossetti and Carboni, 1995; Rossetti et al, 1999). In animal models, it has been shown that acute overstimulation of post-synaptic ionotropic Glu receptors results in excitotoxic neuronal death by disruption of cation and water homeostasis, with chronic overstimulation leading to apoptosis and increased oxidative stress (Coyle and Puttfarcken, 1993). Thus, it has often been suggested (eg, Tsai and Coyle, 1998) that NMDAR-based excitotoxicity underlies the neuronal death and large-scale cortical volume deficits observed in chronic alcoholics (Fein et al, 2002).
In addition to its role in ethanol toxicity, glutamatergic receptor alteration, secondary to ethanol exposure, may have a causal role in the development of alcohol addiction (De Witte, 2004; Szumlinski et al, 2007; Zhu et al, 2007). Ongoing research on small animal and tissue models of alcohol use disorder (AUD) provides ample evidence for the involvement of glutamatergic mechanisms in the development of both deficits and addiction. Until recently, the role of Glu in human AUD has most often been assessed indirectly through analysis of the effects of glutamatergic antagonists on symptoms of withdrawal and intoxication (eg, Bisaga and Popik, 2000).
Proton magnetic resonance spectroscopy (1H-MRS), a technique capable of measuring neurometabolite concentrations non-invasively, provides an ideal tool to directly investigate Glu concentrations in AUD. Numerous studies have used 1H-MRS in human alcohol research (Meyerhoff and Durazzo, 2008). To date, most 1H-MRS studies have focused on the detection of chemical markers of neuronal degradation (Jagannathan et al, 1996; Schweinsburg et al, 2001; Durazzo et al, 2004) and detection of ethanol itself (Mendelson et al, 1990; Meyerhoff et al, 1996; Fein and Meyerhoff, 2000). To our knowledge, only two studies have reported 1H-MRS data on Glu in a chronic AUD population. One study reported no difference in Glx (a combined measure of Glu and glutamine (Gln)) between 12 recently abstinent (1 month) AUD individuals and eight control subjects (Mason et al, 2006). Another study reported no differences between 13 recently abstinent (2 weeks) AUD men and 18 healthy controls in Glu concentrations in the anterior cingulate gyrus or in the insula (Lee et al, 2007). However, in the latter study, a higher ratio of Glu to total creatine was found in the abstinent AUD group and this ratio, along with Glu, was correlated with memory function. None of the study assessed Gln concentrations.
In the present study, we sought to compare metabolic alterations in both Glu and Gln in actively drinking AUD participants, abstinent AUD participants, and healthy controls using single-voxel 1H-MRS. We also investigated relationships of Glu and Gln with scores on the Drinker Inventory of Consequences (DrInC; Miller et al, 1995), a scale reflecting lifetime consequences of alcohol abuse, and on the Form-90, a scale quantifying the frequency and extent of the last 90 days of abuse.
SUBJECTS AND METHODS
Participants
Among the 17 alcohol-dependent participants recruited, 10 met criteria for active AUDs, and seven were in full remission from alcohol use disorders (AUD-R) for at least 1 year. These participants were compared with a sample of 23 healthy controls recruited from the community. All procedures were conducted with the approval of the Human Research and Review Committee at the University Of New Mexico School Of Medicine. All participants provided informed consent. Basic demographic information on each group is provided in Table 1. General inclusion criteria for the study were as follows: (1) willingness to participate in all study components; (2) ability to provide informed consent; (3) ability to read, speak, and understand English at the sixth grade level; (4) ability to provide at least one contact person to assist with collateral interviews; (5) age between 21 and 45 years; (6) at least 48 h after last drink; and (7) a urine sample, free of the presence of cocaine, hallucinogens, barbiturates, benzodiazepines, and opiates.
General exclusion criteria for the study were as follows: (1) history of neurological disorder or disease; (2) history of traumatic brain injury with loss of consciousness for more than 5 min; (3) mental retardation, dementia, or other cognitive impairment of sufficient severity to render the individual incapable of providing informed consent; or (4) suicide attempt in the previous 6 months or current suicidal ideation. As part of a brief health screening interview, self-report of abnormal liver enzyme level or diagnosis of hepatic disease was noted. One participant with a reported history of abnormal liver enzymes was included in the AUD-R group.
The AUD and AUD-R groups were recruited from the University of New Mexico's Alcohol and Substance Abuse Program and Center for Alcoholism, Substance Abuse and Addictions (CASAA). Diagnoses were established using the Structured Clinical Interview for DSM Disorders (SCID), AUD inclusion criteria were: (1) diagnosis of alcohol abuse or dependence active within the past 1 month (ie, not in early or sustained full remission) and (2) two or more days of heavy drinking (five or more drinks for per occasion for a man, four or more drinks per occasion for a woman) in the last 30 days before screening. The AUD-R group met diagnostic criteria for lifetime alcohol dependence in sustained full remission, ie, not active in the previous year. Specific AUD and AUD-R exclusion criteria were: (1) presence of Axis-I schizophrenia spectrum disorders or (2) a first-degree relative with schizophrenia or other psychotic disorder. Healthy control participants were recruited through advertisements in local newspapers or from postings around the Albuquerque area. Control group members reported no history of substance abuse or dependence, with the exception of nicotine, or other Axis-I psychopathology upon interview.
Procedures
Study procedures were conducted at the CASAA and the Mind Research Network in Albuquerque, New Mexico. Clinical interviews typically took place 0–5 days before imaging. AUD and AUD-R participants were administered the DrInC (Miller et al, 1995) as a measure of negative consequences from drinking. DrInC total score was considered here as an estimate of long-term severity of alcohol abuse. Recent consumption data for alcohol and other substances were collected using the Form-90 (Miller and Del Boca, 1994), a timeline follow-back interview in which the participant reported his or her use of alcohol and other substances starting at 90 days preceding the most recent drink to the present. Drinks per drinking day (DPDD) and percentage days drinking (PDD) were chosen as the alcohol variables of interest in order to capture both the intensity and frequency of recent drinking.
Magnetic resonance imaging and MRS data acquisition
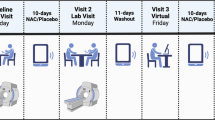
Magnetic resonance imaging and 1H-MRS were performed on a Siemens 3-Tesla TrioTIM scanner using the 12-channel radiofrequency head coil. T1-weighted images were collected in the sagittal plane using a five-echo 3-D MPRAGE sequence (TR/echo time (TE)/TI=2530/1.64, 3.5, 5.36, 7.22, 9.08/1200 ms, flip angle=7°, field of view=256 × 256 mm, matrix=256 × 256, 1 mm thick slice, 192 slices, GRAPPA acceleration factor=2). Using these images, a single 1H-MRS voxel was positioned in the bilateral medial frontal cortex directly superior to the corpus callosum, containing anterior cingulate, middle frontal, and superior frontal gyri (Figure 1). A point-resolved spectroscopy sequence (TR/TE=1.5 s/40 ms, voxel size=20 × 30 × 20 mm, averages=192) was collected, using an TE of 40 ms for improved detection of Glu (Mullins et al, 2008). An unsuppressed water sequence for use as a concentration reference and eddy current correction in post processing was collected with 16 averages and otherwise identical parameters for each single-voxel spectrum.

Placement of 1H-MRS voxel in medial frontal/cingulate cortex.
MRS data analysis
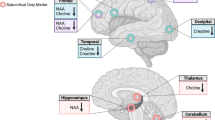
Raw time-domain 1H-MRS data from 4.0 to 1.0 p.p.m. in the spectral dimension were analyzed using LCModel (Provencher, 2001) with the unsuppressed waterscan as a concentration reference. Parameterized macromolecule intensities were included over the fitted spectral region (the LCModel macromolecule intensity set MM20). As a quality assurance measure, LCModel produces a Cramer-Rao lower bound (CRLB) of the fit to the peak of interest. If this value was greater than 20%, the fit was deemed unreliable and excluded from analysis. Metabolite concentrations in molality units of mmol/kg of tissue water were computed for total creatine plus phosphocreatine (Cre), total choline-containing compounds (Cho), myo-inositol (Ins), total N-acetylaspartate plus N-acetyl-aspartylglutamate (NAA), Glu, and Gln. T1-weighted images were segmented into gray matter, white matter, and cerebrospinal fluid (CSF) using SPM5. To calculate tissue and CSF fraction within the spectroscopic voxel, the spatial coordinates of the voxel and T1-weighted image were used to register the voxel volume to the segmentation maps generated from the T1-weighted image. Once this was performed, the gray matter, white matter, and CSF pixels from the segmentation maps that were included in the voxel volume were summed and normalized by the total number of pixels in the volume to arrive at the gray matter, white matter, and CSF fractions in the voxel. Metabolite concentrations were then computed, correcting for partial volume and T1 and T2 relaxation effects using methods described previously (Gasparovic et al, 2006). Figure 2 shows a representative spectrum from a control participant.

An example of a single control subject's spectra, with major metabolites labeled.
Statistical analyses
All statistical analyses were carried out using IBM SPSS, Version 16.0.1. χ2-tests were used to investigate the possible group differences on demographic variables. An omnibus ANOVA was used to test the group effect, with neurometabolites entered as dependent variables. Post-hoc t-tests using a Bonferroni adjustment were then applied to identify which group differences accounted for identified omnibus effects. Relationships between substance use and metabolite levels were explored using bivariate correlation and linear regression.
RESULTS
Demographic information for participants whose data were retained for Glu and Gln analyses (see below) is presented in Table 1. Controls were somewhat younger than the AUD and AUD-R groups, though this difference did not reach the level of statistical significance in the current sample (p=0.31). A greater proportion of control participants were female, but a χ2-analysis showed this difference to be nonsignificant (p=0.36). The control group had significantly more education than AUD and AUD-R groups (p<0.001), who did not differ from each other (p=0.85). The SCID gave evidence of extensive lifetime comorbid psychopathology in AUD and AUD-R groups. In the healthy control group, two participants met criteria for lifetime nicotine dependence. In the alcohol use groups, criteria were met for lifetime diagnosis of bipolar disorder (n=1), major depression (n=7), dysthymia (n=3), brief psychotic disorder (n=1), sedative abuse/dependence (n=1), cannabis abuse/dependence (n=7), stimulant abuse/dependence (n=5), opioid abuse/dependence (n=2), cocaine abuse/dependence (n=9), hallucinogen abuse/dependence (n=3), panic disorder (n=3), agoraphobia (n=1), social phobia (n=3), specific phobia (n=1), obsessive compulsive disorder (n=1), post-traumatic stress disorder (n=3), generalized anxiety disorder (n=7), and nicotine abuse/dependence (n=12). One AUD-R participant reported a history of abnormal liver enzymes and had the lowest Glu level in the sample, but it was within two SDs of the mean.
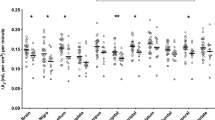
MRS measures for each group are provided in Table 2. For all neurometabolites except Gln, good quality spectra were obtained for every participant. Gln values meeting the CRLB threshold of 20% were obtained for 17 of 23 (74%) controls and 13 of 17 (76%) of the AUD and AUD-R participants, leaving 30 subjects for the analyses. A one-way ANOVA, with Group as the independent variable and Gln and Glu as dependent variables, revealed significant group differences in both Gln (F(92, 30)=5.052, p=0.01), and Glu (F(92, 30)=3.48, p=0.05). For Gln, post-hoc analysis using Bonferroni correction for all possible comparisons revealed significantly lower Gln levels in Controls than in AUD (p=0.03) and marginally lower Gln in Controls than in AUD-R (p=0.08). Gln concentration did not differ between AUD and AUD-R (p=0.99). Using a standard power calculator (Lenth, 2006–2009), it was determined that a mean difference of 1.588 in Gln level between AUD and AUD-R would be necessary to detect a significant difference with the current group sizes, a power of 0.6, and an alpha level set at 0.05 in a two-tailed comparison. For Glu, post-hoc analysis using Bonferroni correction for all possible comparisons revealed higher Glu levels in Controls than in AUD-R (p=0.04), but no significant difference between Controls and AUD (p=0.54). AUD and AUD-R groups showed no difference on Glu concentration (p=0.77). Using a standard power calculator (Lenth, 2006–2009), it was determined that a mean difference of 1.301 in Glu level between AUD and AUD-R would be necessary to detect a significant difference with current group sizes, a power of 0.6, and an alpha level set at 0.05 in a two-tailed comparison.
Effects for sex, age, education, and voxel gray matter concentration were explored by entering each separately into the ANOVA as covariates; none approached the level of significance. Although no group differences were hypothesized, NAA (p=0.29), Cho (p=0.19), Cre (p=0.69), and Ins (p=0.31) metabolite levels were tested using a post-hoc Bonferroni correction in a one-way ANOVA. Again, none approached the level of significance.
To test the extent to which AUD severity was related to Glu–Gln perturbation, lifetime DrInC score was regressed on those metabolite values. The overall regression was significant (R-squared=0.42, p=0.02), with greater severity (higher DrInC score), predicting lower Glu level (β=−0.44, p=0.05) and higher Gln level (β=0.47, p=0.04). Although relationships were not predicted for recent alcohol use, Form-90 DPDD and PDD measures were tested in a similar model, and no values approached the level of significance.
In a number of recent studies, cigarette smoking has been shown to have an independent effect upon neurometabolite levels (Licata and Renshaw, 2010; Mason et al, 2006; Durazzo et al, 2004). To test whether smoking had an effect independent of alcohol use in the present data, Glu and Gln were considered as dependent variables in separate linear regressions. Form-90 estimates of cigarettes smoked per day, number of days smoking, and lifetime weeks of smoking were considered (along with DrInC score) as independent predictors in separate regressions. None of the Glu regressions approached the level of statistical significance. Within the set of regressions for Gln, one approached the level of overall significance. When Gln was regressed on DrInC score and days of cigarette smoking, the regression was marginally significant overall (R-squared=0.363, F=0.132, p=0.084), with an independent effect for DrInC score (β=0.707, p=0.033) and an effect approaching significance at the 0.05 level for days of cigarette smoking (β=−0.523, p=0.098). Hence, there is weak evidence for an independent effect of frequency of cigarette smoking; the effect is opposite to that of alcohol, with more frequent smokers having lower Gln levels.
Post-hoc bivariate correlational analyses adjusted for familywise error were used to investigate relationships between DrInC score and other neurometabolite values (NAA, Ins, Cre), with none approaching the level of significance. When DrInC score was considered, with respect to gray and white matter volumes corrected for familywise error, higher DrInC score predicted reduced voxel gray matter volume (r(17)=−0.61, p=0.04).
DISCUSSION
Lower Glu and higher Gln levels were identified in the anterior cingulate gyrus in young adults with active AUD and with AUD in sustained full remission of at least 1 year, relative to a healthy control group. Hence, lower Glu levels were associated with higher levels of Gln. The extent of abnormality in these metabolites was negatively correlated with the degree of reported lifetime consequences of drinking, interpreted here as the severity of lifetime alcohol use disorder for each subject.
Although glutamatergic hyperexcitability is a cause of withdrawal symptoms and neural death associated with abrupt withdrawal from chronic alcohol exposure (Tsai and Coyle, 1998), abnormal extracellular levels of Glu have been measured in animals long after the acute stage of withdrawal (Rossetti and Carboni, 1995; Dahchour and De Witte, 2003). Glu released from synapses is either transiently bound to NMDA or other receptor sites or is rapidly taken up by excitatory amino acid transporters, located pre- and post synaptically on neurons or astrocytes (reviewed in Gass and Olive, 2008). One of the primary fates of Glu taken up by astrocytes is to be converted to Gln by Gln synthetase before returning to the presynaptic neuron for conversion back into Glu (Magistretti and Pellerin 1999). However, this cycle is not strictly stoichiometric, and there are several other pathways open to astrocytic or neuronal Glu not directly involved in neurotransmission, including conversion into glutathione and entrance into the energy cycle (reviewed by McKenna, 2007).
Upregulation of excitatory transmission has received much attention in the context of alcohol research (eg, Tsai and Coyle, 1998), and a recent study has shown that treatment with acamprosate, a possible NMDR modulator, decreases the Glu/Cre ratio in the anterior cingulate gyrus of AUD patients in early remission (Umhau et al, 2010). Our results, however indicate lower Glu concentrations in AUD subjects without treatment relative to control subjects. It is not clear why a chronic upregulation of excitatory neurotransmission should be accompanied by a decrease in observed Glu concentrations. An alternative possibility is that, given the higher extracellular levels of Glu during withdrawal shown in animal models, there might be a benefit of converting less Gln back into Glu, thereby, diverting a potentially excitotoxic agent into a non-excitotoxic metabolite. The observed shift toward maintaining a larger pool of Gln and smaller pool of Glu may, thus, represent a neuroprotective adaptation. However, this conjecture is entirely speculative, and studies on animal models of AUD, such as experiments measuring altered activities of enzymes in the Gln–Glu cycle, would be necessary to support it. It also is important to bear in mind that 1H-MRS does not distinguish between intracellular (cytosolic vs vesicular or neuronal vs glial) and extracellular pools of Glu. Interestingly, the largest effects in the current study were for Gln concentrations, which may be a more accurate index of overall glutamatergic neurotransmission than Glu (Théberge et al, 2002; Rowland et al, 2005; Ongur et al, 2008). Elevated Gln/Glu ratios have been observed in first-episode schizophrenia (Bustillo et al, 2011), and NMDAR abnormalities (hypofunction) are believed to be important in the pathophysiology of schizophrenia.
The results of the present study suggest that the Glu–Gln cycle remains abnormally long after alcohol cessation, and thus may represent either a chronic effect of alcohol dependence or a predisposing factor for susceptibility to alcohol dependence. In this data set, Gln and Glu levels, as well as voxel gray matter volume were significantly correlated with the accumulated lifetime consequences of drinking. This finding does not establish a causal role for severity of alcohol use in brain abnormality, but future research may pinpoint these measures as direct reflections of chronic exposure to neurotoxic levels of alcohol.
Weaknesses of this study include a small sample size, which particularly limits any conclusions regarding the effects of cigarette smoking or other comorbid diagnoses on neurometabolite levels. Cigarette smoking was confounded with AUD status in this sample, making it impossible to conclusively verify the source of abnormality. Smoking is known to exert neurotoxic effects, and the present analyses indicated a marginally significant, independent effect of smoking beyond that of alcohol abuse. In addition, relative inequalities in gender and age distribution across groups limited our ability to test the effects (or lack thereof) of those variables conclusively. Regarding health status, it is possible to have elevated levels of ammonia or other manifestations of liver damage without the patient's awareness of the condition. As the determination of liver abnormality was based only on the self-report of the subjects, some uncertainty remains regarding the extent to which effects noted herein may be secondary to early stages of liver disease. Finally, we note that the intensities of both Glu and Gln fall in crowded regions of the 1H-MRS spectrum, in which they overlap not only with each other but also with other metabolite and macromolecule intensities. This overlap makes resolving Glu and Gln intensities challenging and dependent on experimental factors, such as spectral line width and the signal-to-noise ratio, as well as several data processing factors, such as adequate modeling of the expected spectral contributions, including macromolecules, and baseline artifacts.
In summary, the results of this study suggest a perturbation of the Gln–Glu cycle in chronic alcoholism, the degree of which correlates with the negative consequences of alcohol abuse. Further studies, particularly human longitudinal studies and animal models, are recommended to elucidate the precise nature and mechanisms of this perturbation.
References
American Psychiatric Association (1994). Diagnostic and Statistical Manual of Mental Disorders: 4th edn. American Psychiatric Association: Washington, DC.
Bisaga A, Popik P (2000). In search of a new pharmacological treatment for drug and alcohol addiction: N-methyl-D-aspartate (NMDA) antagonists. Drug and Alcohol Depend. 59: 1–15.
Bustillo JR, Chen H, Gasparovic C, Mullins P, Caprihan A, Qualls C et al (2011). Glutamate as a marker of cognitive function in schizophrenia: a proton spectroscopic imaging study at 4 Tesla. Biol Psychiatry 69: 19–27.
Coyle JT, Puttfarcken P (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689–695.
Dahchour A, De Witte P (2003). Effects of acamprosate on excitatory amino acids during multiple ethanol withdrawal periods. Alcohol Clin Exp Res 27: 465–470.
De Witte P (2004). Imbalance between neuroexcitatory and neuroinhibitory amino acids causes craving for ethanol. Addict Behav 29: 1325–1339.
Durazzo TC, Gazdzinski S, Bayns P, Meyerhoff DJ (2004). Cigarette smoking exacerbates chronic alcohol-induced brain damage: a preliminary metabolite imaging study. Alcohol Clin Exp Res 28: 1849–1860.
Frye G, Fincher A (2000). Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons. Br J Pharmacol 129: 87–94.
Fein G, Meyerhoff DJ (2000). Ethanol in human brain by magnetic resonance spectroscopy: correlation with blood and breath levels, relaxation, and magnetization transfer. Alcohol Clin Exp Res 24: 1227–1235.
Fein G, Sclafani V, Cardenas VA, Goldman H, Tolou-Shams M, Meyerhoff DJ (2002). Cortical gray matter loss in treatment-naive alcohol dependent individuals. Alcohol Clin Exp Res 26: 558–564.
Gasparovic C, Song T, Devier D, Bockholt HJ, Caprihan A, Mullins P et al (2006). Use of tissue water as a concentration reference for proton spectroscopic imaging. Magn Reson Med 55: 1219–1226.
Gass J, Olive M (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 75: 218–265.
Hoffman PL (1995). Glutamate receptors in alcohol withdrawal-induced neurotoxicity. Metab Brain Dis 10: 73–79.
Jagannathan NR, Desai NG, Raghunathan P (1996). Brain metabolite changes in alcoholism: an in vivo proton magnetic resonance spectroscopy (MRS) study. Magn Reson Imaging 14: 553–557.
Lee E, Jang DP, Kim JJ, Kyoon AS, Sangjin P, In-Young K et al (2007). Alteration of brain metabolites in young alcoholics without structural changes. Neuroreport 18: 1511–1514.
Lenth RV (2006–2009). Java applets for power and sample size [computer software]. Retrieved 16 December 2010 from http://www.stat.uiowa.edu/∼rlenth/Power.
Licata SC, Renshaw PF (2010). Neurochemistry of drug action: insights from proton magnetic resonance spectroscopic imaging and their relevance to addiction. Ann N Y Acad Sci 1187: 148–171.
Lovinger DM (1993). High ethanol sensitivity of recombinant AMPA-type glutamate receptors expressed in mammalian cells. Neurosci Lett 159: 83–87.
Magistretti PJ, Pellerin L (1999). Astrocytes couple synaptic activity to glucose utilization in the brain. News Physiol Sci. 14: 177–182.
Mason GF, Petrakis IL, de Graaf RA, Gueorguieva E, Coric V, Epperson CN et al (2006). Cortical gamma-amino butyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry 59: 85–93.
McKenna M (2007). The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res 58: 3347–3358.
Mendelson JH, Woods BT, Chiu TM, Mello NK, Lukas SE, Teoh SK et al (1990). In vivo proton magnetic resonance spectroscopy of alcohol in human brain. Alcohol 7: 443–447.
Meyerhoff DJ, Durazzo TC (2008). Proton magnetic resonance spectroscopy in alcohol use disorders: a potential new endophenotype? Alcohol Clin Exp Res 32: 1146–1158.
Meyerhoff DJ, Rooney WD, Tokumitsu T, Weiner MW (1996). Evidence of multiple ethanol pools in the brain: an in vivo proton magnetization transfer study. Alcohol Clin Exp Res 20: 1283–1288.
Miller WR, Del Boca FC (1994). Measurement of drinking behavior using the Form 90 family of instruments. J Stud Alcohol Suppl 12: 112–118.
Miller WR, Tonigan JS, Longabaugh R (1995). The Drinker Inventory of Consequences (DrInC): An Instrument for Assessing Averse Consequence of Alcohol Abuse. Project MATCH Monograph Series: Rockville, MD.
Mullins PG, Chen H, Xu J, Caprihan A, Gasparovic C (2008). Comparative reliability of proton spectroscopy techniques designed to improve detection of J-coupled metabolites. Magn Reson Med 60: 964–969.
Ongur D, Jensen J, Prescot A, Stork C, Lundy M, Cohen B et al (2008). Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry 64: 718–726.
Provencher SW (2001). Automatic quantitation of localized in vivo1H spectra with LCModel NMR in Biomedicine, Special Issue: NMR Spectroscopy Quantitation 14: 260–264.
Rossetti Z, Carboni S, Fadda F (1999). Glutamate-induced increase of extracellular glutamate through N-methyl-D-aspartate receptors in ethanol withdrawl. Neuroscience 93: 1135–1140.
Rossetti ZL, Carboni S (1995). Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur J Pharmacol 283: 177–183.
Rourke S, Loberg I (1996). Neurobehavioral correlates of alcoholism. In: Grant I, Adams K (eds). Neuropsychological Assessment of Neuropsychiatric Disorders. Oxford, New York.
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E et al (2005). Effects of ketamine in anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry 162: 394–396.
Schweinsburg BC, Taylor MJ, Alhassoon OM, Videen JS, Brown GG, Patterson TL et al (2001). Chemical pathology in brain white matter of recently detoxified alcoholics: a 1H magnetic resonance spectroscopy investigation of alcohol-associated frontal lobe injury. Alcohol Clin Exp Res 25: 924–934.
Szumlinski KK, Diab ME, Friedman R, Henze L, Lominac K, Bowers S (2007). Accumbens neurochemical adaptations produced by binge-like alcohol consumption. Psychopharmacology 190: 415–431.
Théberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J et al (2002). Glutamate and glutamine measured with 4.0 T Proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry 159: 1944–1946.
Tsai G, Coyle J (1998). The role of glutamatergic neurotransmission in the pathophysiology of alcoholism. Annu Rev Med 49: 173–184.
Umhau JC, Momenan R, Schwandt ML, Singley E, Lifshitz M, Doty L et al (2010). Effect of acamprosate on magnetic resonance spectroscopy measures of central glutamate in detoxified alcohol-dependent individuals: a randomized controlled experimental medicine study. Arch Gen Psychiatry 67: 1069–1077.
US Department of Health and Human Services, Substance Abuse and Mental Health Services Administration (2002). Results from the 2001 National Household Survey on Drug Abuse: Volume I. Summary of National Findings (Office of Applied Studies, NHSDA Series H-17 ed.) (BKD461, SMA 02-3758) US Government Printing Office: Washington, DC.
Yeo RA, Gasparovic C, Merideth F, Ruhl D, Doezema D, Mayer AR (2011). A longitudinal proton magnetic resonance spectroscopy study of mild traumatic brain injury. J Neurotrauma 28: 1–11.
Zhu W, Bie B, Pan ZZ (2007). Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci 27: 289–298.
Acknowledgements
This paper was supported by grants to Dr Thoma (PI: K23AA016544 & R21AA0173134) from the National Institute on Alcohol Abuse and Alcoholism and by funding from the Mind Research Network (DE-FG02-99ER62764).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Thoma, R., Mullins, P., Ruhl, D. et al. Perturbation of the Glutamate–Glutamine System in Alcohol Dependence and Remission. Neuropsychopharmacol 36, 1359–1365 (2011). https://doi.org/10.1038/npp.2011.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.20
